National Cancer Institute
Post Date: Apr 3, 2024
Genetics of Breast and Gynecologic Cancers includes the hereditary cancer syndromes BRCA1/BRCA2 (hereditary breast and ovarian cancer), Lynch syndrome, Li Fraumeni syndrome, ATM, PALB2, CHEK2 and other genes. Get comprehensive information on these syndromes in this clinician summary.
Genetics of Breast and Gynecologic Cancers
Executive Summary
This executive summary reviews the topics covered in this PDQ summary on the genetics of breast and gynecologic cancers.
- Inheritance and Risk
Factors suggestive of a genetic contribution to both breast cancer and gynecologic cancer include 1) an increased incidence of these cancers among individuals with a family history of these cancers; 2) multiple family members affected with these and other cancers; and 3) a pattern of cancers compatible with autosomal dominant inheritance. Both males and females can inherit and transmit an autosomal dominant cancer predisposition gene.
Additional factors coupled with family history can influence an individual's risk of developing cancer—such as reproductive history, contraceptive and hormone replacement use, radiation exposure early in life, alcohol consumption and smoking, and physical activity.
Risk assessment models have been developed to clarify an individual's 1) lifetime risk of developing breast and/or gynecologic cancer; 2) likelihood of having a pathogenic variant in BRCA1 or BRCA2; and 3) likelihood of having a pathogenic variant in one of the mismatch repair genes associated with Lynch syndrome.
- Associated Genes and Syndromes
Breast and ovarian cancer are present in several autosomal dominant cancer syndromes, although they are most strongly associated with highly penetrant germline pathogenic variants in BRCA1 and BRCA2. Other genes, such as PALB2, TP53 (associated with Li-Fraumeni syndrome), PTEN (associated with PTEN hamartoma tumor syndromes, including Cowden syndrome), CDH1 (associated with diffuse gastric and lobular breast cancer syndrome), and STK11 (associated with Peutz-Jeghers syndrome), confer a risk to either or both of these cancers with relatively high penetrance.
Inherited endometrial cancer is most commonly associated with Lynch syndrome, a condition caused by inherited pathogenic variants in the highly penetrant mismatch repair genes MLH1, MSH2, MSH6, PMS2, and EPCAM. Colorectal cancer (and, to a lesser extent, ovarian cancer and stomach cancer) is also associated with Lynch syndrome.
CHEK2, BRIP1, RAD51C, RAD51D, and ATM are moderate penetrance genes that are associated with increased breast and/or gynecologic cancer risk. Genome-wide searches are showing promise in identifying common, low-penetrance susceptibility alleles for many complex diseases, including breast and gynecologic cancers, but the clinical utility of these findings remains uncertain.
- Clinical Management
Breast cancer screening strategies, including breast magnetic resonance imaging and mammography, are commonly performed in carriers of BRCA pathogenic variants and in individuals at increased risk of breast cancer. Initiation of screening is generally recommended at earlier ages and at more frequent intervals in individuals with an increased risk due to genetics and family history than in the general population. There is evidence to demonstrate that these strategies have utility in early detection of cancer. In contrast, there is currently no evidence to demonstrate that ovarian cancer screening using cancer antigen–125 testing and transvaginal ultrasound leads to early detection of cancer.
Risk-reducing surgeries, including risk-reducing mastectomy (RRM) and risk-reducing salpingo-oophorectomy (RRSO), have been shown to significantly reduce the risk of developing breast and/or ovarian cancer and improve overall survival in carriers of BRCA1 and BRCA2 pathogenic variants. Chemoprevention strategies for breast cancer and chemoprevention strategies for ovarian cancer have been examined in this population. For example, tamoxifen use has been shown to reduce the risk of contralateral breast cancer among carriers of BRCA1 and BRCA2 pathogenic variants after treatment for breast cancer, but there are limited data in the primary cancer prevention setting to suggest that it reduces the risk of breast cancer among healthy female carriers of BRCA2 pathogenic variants. The use of OCs also has been associated with a protective effect on the risk of developing ovarian cancer, including in carriers of BRCA1 and BRCA2 pathogenic variants, with no association of increased risk of breast cancer when using formulations developed after 1975.
- Psychosocial and Behavioral Issues
Psychosocial factors influence decisions about genetic testing for inherited cancer risk and risk-management strategies. Uptake of genetic testing varies widely across studies. Psychological factors that have been associated with testing uptake include cancer-specific distress and perceived risk of developing breast or ovarian cancer. Studies have shown low levels of distress after genetic testing for both carriers and noncarriers, particularly in the longer term. Uptake of RRM and RRSO also varies across studies and may be influenced by factors such as cancer history, age, family history, recommendations of the health care provider, and pretreatment genetic education and counseling. Patients' communication with their family members about an inherited risk of breast and gynecologic cancer is complex; gender, age, and the degree of relatedness are some elements that affect disclosure of this information. Research is ongoing to better understand and address psychosocial and behavioral issues in high-risk families.
Introduction
General Information
Among women in the United States, breast cancer is the most commonly diagnosed cancer after nonmelanoma skin cancer, and it is the second leading cause of cancer deaths after lung cancer. In 2024, an estimated 313,510 new cases of breast cancer (including 2,790 cases in men) will be diagnosed, and 42,780 deaths (including 530 deaths in men) will occur. The incidence of breast cancer, particularly for estrogen receptor (ER)–positive cancers occurring after age 50 years, is declining and has declined at a faster rate since 2003. This may be temporally related to a decrease in hormone replacement therapy (HRT) after early reports from the Women’s Health Initiative (WHI). An estimated 19,680 new cases of ovarian cancer are expected in the United States in 2024, with an estimated 12,740 deaths. Ovarian cancer is the sixth most deadly cancer in women. An estimated 67,880 new cases of endometrial cancer are expected in the United States in 2024, with an estimated 13,250 deaths. (Refer to the PDQ summaries on Breast Cancer Treatment; Ovarian Epithelial, Fallopian Tube, and Primary Peritoneal Cancer Treatment; and Endometrial Cancer Treatment for more information about breast, ovarian, and endometrial cancer rates, diagnosis, and management.)
A possible genetic contribution to both breast and ovarian cancer risk is indicated by the increased incidence of these cancers among women with a family history (refer to the Risk Factors for Breast Cancer, Risk Factors for Ovarian Cancer, and Risk Factors for Endometrial Cancer sections below for more information), and by the observation of some families in which multiple family members are affected with breast and/or ovarian cancer, in a pattern compatible with an inheritance of autosomal dominant cancer susceptibility. Formal studies of families (linkage analysis) have subsequently proven the existence of autosomal dominant predispositions to breast and ovarian cancer and have led to the identification of several highly penetrantgenes as the cause of inherited cancer risk in many families. (Refer to the PDQ summary Cancer Genetics Overview for more information about linkage analysis.) Pathogenic variants in these genes are rare in the general population and are estimated to account for no more than 5% to 10% of breast and ovarian cancer cases overall. It is likely that other genetic factors contribute to the etiology of some of these cancers.
Risk Factors for Breast Cancer
This section discusses factors that can modify an individual's risk of developing breast cancer. These risk factors can affect women in the general population, women who have a family histories of breast cancer, and women who carry pathogenic variants in breast cancer risk genes. For more information on breast cancer risk factors in the general population, see Breast Cancer Prevention, and for more information on risks associated with BRCA1/2 pathogenic variants, see theCancer Risks, Spectrum, and Characteristics section in BRCA1 and BRCA2: Cancer Risks and Management.
The following breast cancer risk factors are discussed in this section:
- Age.
- Family history of breast cancer.
- Benign breast disease, mammographic density, and background parenchymal enhancement.
- Parity, age at first birth, and breastfeeding.
- Contraceptives.
- Hormone replacement therapy (HRT).
- Radiation exposure.
- Alcohol and smoking.
- Physical activity.
These factors can increase or decrease breast cancer risk in all women. However, they may affect breast cancer risk differently in women with increased breast cancer susceptibility (i.e., women who have high-risk family histories and/or pathogenic variants in hereditary breast cancer genes). Factors that increase breast cancer risk in the general population may lower breast cancer risk, increase breast cancer risk more than expected, or have no effect on breast cancer risk in women with high breast cancer susceptibility. In some cases, these risk factors may affect high-risk women in the same way that they affect average-risk women. Furthermore, modifying risk factors has a greater effect on the absolute breast cancer risk in women with high breast cancer susceptibility than in women with low breast cancer susceptibility. It is imperative that providers discuss breast cancer risk factors with high-susceptibility patients, since risk patterns deviate from those seen in women in the general population. Providers may also want to convey whether these risk factors increase, decrease, or do not affect breast cancer risk in women with high breast cancer susceptibility, based on available evidence. This information may change how providers approach breast cancer risk management in women with high breast cancer susceptibility.
Age
Like other cancer types, breast cancer's cumulative risk increases with age. As individuals age, they encounter more environmental exposures and accumulate genomic changes. Hence, most breast cancers occur after age 50 years. Women with pathogenic variants in breast cancer risk genes often develop breast cancer at younger ages than women with sporadic breast cancers.
Family history of breast cancer
A family history of breast cancer is a well-established, consistent risk factor for breast cancer. Approximately 5% to 10% of women with breast cancer also had a mother or sister with breast cancer in cross-sectional studies. About 10% to 20% of women had a first-degree relative (FDR) or a second-degree relative (SDR) with breast cancer. A pooled analysis of 38 studies showed that women had increased breast cancer risk when they had at least one FDR with breast cancer (relative risk [RR], 2.1; 95% confidence interval [CI], 2.0–2.2). A large population-based study that used the Swedish Family Cancer Database found that women had a significantly increased risk of breast cancer when they had a mother or a sister with breast cancer.
The following factors can increase a woman's breast cancer risk:
- Large number of affected relatives.
- Family members who were diagnosed with breast cancer at young ages.
- Family members with bilateral breast cancers.
- Family members with multiple ipsilateral breast cancers.
- Male relatives with breast cancer.
Furthermore, women with family histories of multiple breast cancers had higher hazard ratios (HRs) (HR, 2.7; 95% CI, 2.6–2.9) than women who had a single breast cancer in their families (HR, 1.8; 95% CI, 1.8–1.9). When women had multiple breast cancers in their families (with one breast cancer occurring before age 40 years), the HR was 3.8 (95% CI, 3.1–4.8). However, breast cancer risk also significantly increased when a relative was diagnosed with breast cancer at 60 years or older, suggesting that having a relative with breast cancer at any age can increase risk. Another study in women with unilateral versus contralateral breast cancer (CBC) evaluated CBC risk among family members. Results indicated that women with at least one affected FDR had an 8.1% chance of developing CBC after 10 years. Participants' risks also increased when relatives were diagnosed with breast cancer before age 40 years (10-year absolute risk [AR], 13.5%; 95% CI, 8.8%–20.8%) or if relatives had CBC (10-year AR, 14.1%; 95% CI, 9.5%–20.7%). These risks were similar to those seen among BRCA carriers (10-year AR, 18.4%; 95% CI, 16.0%–21.3%). These risk estimates remained unchanged when the analysis was restricted to women who tested negative for a pathogenic variant in BRCA1/BRCA2, ATM, CHEK2, or PALB2.
Albright et al. addressed how affected third-degree relatives (TDRs) can contribute to an individual's breast cancer risk. These researchers used the Utah Population Database and the Utah Cancer Registry to estimate RRs for participants to develop breast cancer. They collected family histories with FDRs, SDRs, and TDRs and included both paternal and maternal relatives. They confirmed that individuals with affected FDRs had the highest breast cancer risk, particularly if the FDR was diagnosed with breast cancer early in life. When participants had five or more affected TDRs (and no FDRs/SDRs with breast cancer), they had an RR of 1.32 (95% CI, 1.11–1.57).
One of the largest studies of twins ever conducted examined 80,309 monozygotic twins and 123,382 dizygotic twins. This study had a heritability estimate of 31% for breast cancer (95% CI, 11%–51%). If a monozygotic twin had breast cancer, her twin sister had a 28.1% chance of developing breast cancer (95% CI, 23.9%–32.8%), and if a dizygotic twin had breast cancer, her twin sister had a 19.9% chance of developing breast cancer (95% CI, 17%–23.2%). These estimates suggest that monozygotic twins have a 10% higher risk of developing breast cancer than dizygotic twins. However, the high rate of discordance seen, even between monozygotic twins, suggests that environmental factors can also modify breast cancer risk.
Benign breast disease, mammographic density, and background parenchymal enhancement
Benign breast disease (BBD)
- BBD is a broad group of conditions characterized by non-cancerous changes in breast tissue. BBD can be divided into three categories: nonproliferative lesions, proliferative lesions without atypia, and atypical hyperplasias. BBD is a consistent risk factor for breast cancer in the general population.
- BBD is also an important risk factor in women who have high breast cancer susceptibility due to family histories of cancer or pathogenic variants in breast cancer risk genes. For example, a study of 17,154 women found that women with a history of BBD have an increased risk of breast cancer that is independent of their underlying familial and genetic risks. However, breast cancer risk associated with personal histories of BBD did not vary between women with BRCA1 pathogenic variants (RR, 1.64; 95% CI, 1.04–2.58), women with BRCA2 pathogenic variants (RR, 1.34; 95% CI, 0.78–2.3), and women who only had family histories of breast cancer (RR, 1.31; 95% CI, 1.13–1.53). In women with high breast cancer susceptibility, BBD can further increase breast cancer risk, because it multiplies their underlying familial and genetic risks.
Mammographic density
- Women with dense breast tissue (assessed by mammogram) also have an increased risk of developing breast cancer. Studies have shown that breast density likely has a genetic etiology.
- A systematic review reported that women who had dense breast tissue and an FDR with breast cancer had an increased chance of developing breast cancer. Two retrospective studies also investigated the association between mammographic density and breast cancer risk in BRCA1 and BRCA2 carriers. These retrospective studies had samples of 206 and 691 BRCA pathogenic variant carriers. In these studies, 96 and 248 women developed breast cancer, respectively. The studies found that mammographic density is an independent risk factor for breast cancer in both BRCA1 and BRCA2 pathogenic variant carriers. Associations between breast density and breast cancer risk were similar to those observed in the general population (RR, 2.30 for density ≥50% vs. <50%).
Background parenchymal enhancement (BPE)
- Like breast density (assessed by mammogram), BPE (assessed by breast magnetic resonance imaging [MRI]) may increase breast cancer risk. Data have shown that moderate BPE (odds ratio [OR], 1.6; 95% CI, 1.0–2.6) and mild BPE (OR, 2.1; 95% CI, 1.5–3.0) can increase breast cancer risk in women with high breast cancer susceptibility. However, an association between mild/moderate BPE and breast cancer risk was not found in women with average breast cancer susceptibility.
Parity, age at first birth, and breastfeeding
Parity
- A large prospective study analyzed the relationship between parity and breast cancer risk in female BRCA1 and BRCA2 carriers. Results showed that parity affected breast cancer risk in BRCA1 and BRCA2 carriers differently. Breast cancer risk increased in uniparous BRCA1 carriers and parous BRCA2 carriers. In BRCA1 carriers, there was no overall association between parity and breast cancer risk when compared with nulliparity and breast cancer risk. Uniparous BRCA1 carriers were at an increased risk of breast cancer in the prospective analysis (HRprospective , 1.69; 95% CI, 1.09–2.62) when compared with nulliparous BRCA1 carriers. The results also suggested that uniparous women who breastfed may have decreased breast cancer risk when compared with those who did not breastfeed. In BRCA2 carriers, being parous was associated with a 33% increase in breast cancer risk (HRcombined , 1.33; 95% CI, 1.05–1.69). Multiparity did not decrease breast cancer risk in BRCA2 carriers, unless they had at least four full-term pregnancies (HRcombined, 0.72; 95% CI, 0.54–0.98).
Age at first birth
- In the general population, breast cancer risk increases when women have early menarche and/or late menopause. Breast cancer risk decreases when a woman’s first full-term pregnancy occurs at a young age. However, these risk factors can affect women with high breast cancer susceptibility differently than women in the general population. BRCA1 and BRCA2 pathogenic variant carriers who become pregnant prior to age 30 years may have increased breast cancer risk. This effect is even more significant in BRCA1 pathogenic variant carriers. BRCA1 and BRCA2 pathogenic variant carriers who developed breast cancer during pregnancy or became pregnant after developing breast cancer did not experience adverse survival outcomes.
Breastfeeding
- Breastfeeding can reduce breast cancer risk in BRCA1 (but not BRCA2) pathogenic variant carriers. Breastfeeding for long periods of time was associated with decreased breast cancer risk in BRCA1 carriers (P-trend = .0003).
Reproductive history can also affect a woman's risk for ovarian cancer and endometrial cancer. For more information, see the Risk Factors for Ovarian Cancer and Risk Factors for Endometrial Cancer sections.
Contraceptives
Breast cancer risk is one of the factors to consider when prescribing contraceptives, which assist with pregnancy control, abnormal bleeding, and other gynecological symptoms. Oral contraceptives (OCs) may slightly increase breast cancer risk in long-term users, but this appears to be a short-term effect.
Some studies show that OC use does not further increase breast cancer risk in women with high breast cancer susceptibility. For example, a meta-analysis with data from 54 studies showed that women with family histories of breast cancer did not have increased breast cancer risk from OC use. Although the data are not entirely consistent, a meta-analysis of BRCA1/BRCA2 pathogenic variant carriers concluded that breast cancer risk did not significantly increase when participants used OCs. More specifically, the International BRCA1/2 Carrier Cohort Study (IBCCS), the Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab) Follow-Up Study, and the Breast Cancer Family Registry (BCFR) did not report associations between OC use and increased breast cancer risk in women with BRCA1 pathogenic variants. In fact, OCs are sometimes recommended for ovarian cancer prevention in BRCA1 and BRCA2 pathogenic variant carriers. For more information, see the Oral contraceptives and Risk Factors for Ovarian Cancer sections. However, in the prospective analyses of the IBCCS, kConFab, and BCFR studies mentioned above, women with BRCA2 pathogenic variants had increased breast cancer risk when they took OCs (HR, 1.75; 95% CI, 1.03–2.97). Additionally, a systematic review of the published data concluded that it is unclear if OC use increases breast cancer risk in BRCA1/2 carriers due to inconsistencies across studies.
Some studies also suggest that the year an OC was made and a woman's age when beginning OC use may matter. For example, OCs made before 1975 are associated with increased breast cancer risk in BRCA1/2 carriers (summary relative risk [SRR], 1.47; 95% CI, 1.06–2.04). A case-control study of 2,492 matched pairs of women with a BRCA1 pathogenic variant also found that OC use significantly increased breast cancer risk when women began using OCs prior to age 20 years (OR, 1.45; 95% CI, 1.20–1.75).
Other contraceptive methods have not been studied in women with pathogenic variants in breast cancer risk genes. However, studies have investigated associations between intrauterine devices and breast cancer risk in the general population. A meta-analysis and systematic review of seven studies examined the effect of the levonorgestrel-releasing intrauterine system (LNG-IUS) on breast cancer risk. The meta-analysis included studies that controlled for family history of breast cancer, but associations were not separately evaluated or stratified by family history of breast cancer. In LNG-IUS users, breast cancer risk increased in all women (OR, 1.16; 95% CI, 1.06–1.28), in women younger than 50 years (OR, 1.12; 95% CI, 1.02–1.22), and in women 50 years and older (OR, 1.52; 95% CI, 1.34–1.72).
Hormone replacement therapy
Both observational studies and randomized clinical trials have examined the association between postmenopausal HRT and breast cancer. Short-term use of HRT for treatment of postmenopausal symptoms appears to confer little or no breast cancer risk. A meta-analysis with data from 51 observational studies found a 1.35 RR for breast cancer (95% CI, 1.21–1.49) in women who used HRT for 5 or more years after menopause. The WHI, a randomized, controlled trial of about 160,000 postmenopausal women, investigated the risks and benefits of HRT. The estrogen-plus-progestin arm of the study, in which more than 16,000 women were randomly assigned to receive combined estrogen and progestin or placebo, was halted early because health risks exceeded health benefits. Significant increases in both total breast cancer cases (245 in the estrogen-plus-progestin group vs. 185 in the placebo group) and invasive breast cancer cases (199 in the estrogen-plus-progestin group vs. 150 in the placebo group) prompted early closure of the study (RR, 1.24; 95% CI, 1.02–1.5; P< .001). Risks for coronary heart disease, stroke, and pulmonary embolism also increased in the estrogen-plus-progestin group. The WHI study did not stratify data by participants' family histories of breast cancer, and subjects were not systematically tested for BRCA1/BRCA2 pathogenic variants. Similar findings were seen in the estrogen-progestin arm of the prospective, observational Million Women’s Study in the United Kingdom. However, breast cancer risk was not elevated in women randomly assigned to the estrogen-only group when compared with those in the placebo group in the WHI study (RR, 0.77; 95% CI, 0.59–1.01). Hysterectomy was required for women to qualify for the estrogen-only arm of this study; 40% of these patients also had a bilateral oophorectomy, which can potentially decrease breast cancer risk.
Among women with family histories of breast cancer, the associations between HRT and breast cancer risk have not been consistent. Some studies suggested risk was particularly elevated among women with family histories of breast cancer, while others did not report an interaction between these factors. A large meta-analysis found that women who used HRT had increased breast cancer risk. However, risk did not differ significantly between subjects with or without family histories of cancer.
The effect of HRT on breast cancer risk among carriers of BRCA1 and BRCA2 pathogenic variants has been studied in the context of bilateral risk-reducing oophorectomy. Short-term HRT use does not seem to alter an oophorectomy's protective effect on breast cancer risk. For example, a prospective, longitudinal cohort study recruited BRCA1 carriers from 80 centers in 17 countries. This study found that HRT use after oophorectomy was not associated with increased breast cancer risk in BRCA1 carriers. The HR was 0.97 (95% CI, 0.62–1.52) for individuals who used HRT when compared with individuals who had never used HRT. However, the effects of estrogen-only HRT and estrogen-plus-progesterone HRT differed. After a 10-year follow-up period, the cumulative breast cancer incidence was 12% in women who used estrogen-only HRT and 22% in women who used estrogen-plus-progesterone HRT. These associations were stronger for women who underwent oophorectomy before age 45 years. The study concluded that using estrogen-only HRT after oophorectomy did not increase risk of BRCA1-associated breast cancers. However, the potential harmful effects of progesterone-containing HRT warrant further study. For more information, see the HRT in Carriers of BRCA1/BRCA2 Pathogenic Variants section in BRCA1 and BRCA2: Cancer Risks and Management.
HRT use may also increase a woman's chance of developing endometrial cancer. For more information, see the Hormones section.
Radiation exposure
Radiation exposure can increase an individual's breast cancer risk. This is demonstrated by the survivors of the atomic bombings in Hiroshima and Nagasaki and by women who have received therapeutic radiation treatments to the chest and upper body. However, it is unclear how much radiation exposure affects breast cancer risk in women with high breast cancer susceptibility.
Early data suggested that carriers of BRCA1 and BRCA2 pathogenic variants may have increased sensitivity to radiation, which may contribute to cancer susceptibility. Studies have shown that individuals with germline ATM and TP53 variants also have increased sensitivity to radiation.
It is possible that radiation exposure from diagnostic procedures, including mammography, poses a greater risk to women with high breast cancer susceptibility than to women who are at average risk of developing breast cancer. Therapeutic radiation could also increase cancer risk in women with high breast cancer susceptibility. However, a cohort study of BRCA1 and BRCA2 pathogenic variant carriers treated with breast-conserving therapy did not show evidence of increased radiation sensitivity in participants. Sequelae were not observed in the breasts, lungs, or bone marrow of BRCA carriers.
Conversely, tumors in women with pathogenic variants in breast cancer risk genes may be more responsive to radiation treatment than tumors in women at average breast cancer risk. Studies examining the impact of radiation exposure in carriers of BRCA1 and BRCA2 pathogenic variants have had conflicting results. A large European study showed a dose-response relationship, in which breast cancer risk increased with total radiation exposure. However, this occurred most often when patients had nonmammographic radiation exposure before age 20 years. A significant association was not observed between prior mammography exposure and breast cancer risk in a prospective study of 1,844 BRCA1 carriers and 502 BRCA2 carriers without breast cancer diagnoses upon study entry. The average follow-up period in this study was 5.3 years.
A retrospective cohort study estimated the effect of adjuvant radiation therapy (for primary breast cancer) on CBC risk in BRCA1 and BRCA2 carriers (N, 691; median follow-up period, 8.6 y). An association was not found between radiation therapy and CBC risk (HR, 0.82; 95% CI, 0.45–1.45). This was also true in patients who were younger than 40 years when they were diagnosed with their primary breast cancers (HR, 1.36; 95% CI, 0.60–3.09). A study examined the impact of radiation therapy on CBC risk in ATM, BRCA1/2, and CHEK2 1100delC carriers. CBC risk was not modified by radiation therapy, even though these women had a higher baseline risk of CBC than women in the general population (BRCA1/2 pathogenic variant carriers without radiation therapy: RR, 3.52; 95% CI, 1.76–7.01; BRCA1/2 pathogenic variant carriers with radiation therapy: RR, 4.46; 95% CI, 2.96–6.71). Thus, it is important to differentiate individuals with increased CBC risk due to pathogenic variants from individuals with increased CBC risk due to radiation therapy. For more information, see the Mammography section in BRCA1 and BRCA2: Cancer Risks and Management.
Alcohol and smoking
The risk of breast cancer increases by approximately 10% for each 10 g of daily alcohol intake (approximately one drink or less) in the general population. Prior studies of BRCA1/BRCA2 pathogenic variant carriers have not found an association between alcohol consumption and increased breast cancer risk. The association between cigarette smoking and breast cancer risk in women with BRCA1/2 pathogenic variants is inconclusive.
Recent studies have evaluated the association between alcohol consumption, tobacco smoking, and breast cancer risk in individuals with BRCA1/2 pathogenic variants or family histories of breast cancer. One study evaluated if tobacco smoking and alcohol consumption are associated with increased breast cancer risk in BRCA1 and BRCA2 carriers using pooled data from an international cohort. This study did not find an association between alcohol consumption and increased breast cancer risk in BRCA1 and BRCA2 carriers. Parous BRCA carriers who smoked for more than 5 years before their first full-term pregnancy had a significantly increased breast cancer risk when compared with parous BRCA carriers who did not smoke. A prospective study evaluating a cohort of women with family histories of breast cancer found that alcohol consumption was associated with an increased number of ER-positive breast cancers in women at the lowest quantile of absolute breast cancer risk (HR, 1.46; 95% CI, 1.07–1.99). Cigarette smoking was also associated with increased breast cancer risk in those at the highest quantile of absolute breast cancer risk.
Physical activity
Increased physical activity has been associated with reduced breast cancer risk in most epidemiological studies. This risk reduction has also been seen in studies of female BRCA1 or BRCA2 pathogenic variant carriers. For example, one study reported a 38% reduction in premenopausal breast cancer risk from moderate physical activity (OR for the top quartile of physical activity compared with the lowest level, 0.62; 95% CI, 0.40–0.96). This reduction in breast cancer risk has been seen in women with varying levels of breast cancer susceptibility, including women who have family histories of breast cancer but do not have known BRCA1 or BRCA2 pathogenic variants.
Risk Factors for Ovarian Cancer
Refer to the PDQ summary on Ovarian, Fallopian Tube, and Primary Peritoneal Cancers Prevention for information about risk factors for ovarian cancer in the general population.
Age
Ovarian cancer incidence rises in a linear fashion from age 30 years to age 50 years and continues to increase, though at a slower rate, thereafter. Before age 30 years, the risk of developing epithelial ovarian cancer is remote, even in hereditary cancer families.
Family history including inherited cancer genes
Although reproductive, demographic, and lifestyle factors affect risk of ovarian cancer, the single greatest ovarian cancer risk factor is a family history of the disease. A large meta-analysis of 15 published studies estimated an OR of 3.1 for the risk of ovarian cancer associated with at least one FDR with ovarian cancer.
Reproductive history
Nulliparity is consistently associated with an increased risk of ovarian cancer, including among carriers of BRCA/BRCA2 pathogenic variants, yet a meta-analysis identified a risk reduction only in women with four or more live births. Risk may also be increased among women who have used fertility drugs, especially those who remain nulligravid. Several studies have reported a risk reduction in ovarian cancer after OC use in carriers of BRCA/BRCA2 pathogenic variants; a risk reduction has also been shown after tubal ligation in BRCA1 carriers, with a statistically significant decreased risk of 22% to 80% after the procedure. Breastfeeding for more than 12 months may also be associated with a reduction in ovarian cancer among carriers of BRCA1/BRCA2 pathogenic variants. On the other hand, evidence is growing that the use of menopausal HRT is associated with an increased risk of ovarian cancer, particularly in long-time users and users of sequential estrogen-progesterone schedules.
Surgical history
Bilateral tubal ligation and hysterectomy are associated with reduced ovarian cancer risk, including in carriers of BRCA/BRCA2 pathogenic variants. Ovarian cancer risk is reduced more than 90% in women with documented BRCA1 or BRCA2 pathogenic variants who chose risk-reducing salpingo-oophorectomy (RRSO). In this same population, risk-reducing oophorectomy also resulted in a nearly 50% reduction in the risk of subsequent breast cancer. While some studies have shown more benefit for breast cancer reduction in patients with BRCA2 versus BRCA1 pathogenic variants, others have shown no benefit for BRCA1 carriers. Additionally, many of the studies remain underpowered to demonstrate benefit. (Refer to the Risk-reducing salpingo-oophorectomy for breast cancer risk reduction section in BRCA1 and BRCA2: Cancer Risks and Management for more information about these studies.)
Oral contraceptives (OCs)
Use of OCs for 4 or more years is associated with an approximately 50% reduction in ovarian cancer risk in the general population. A majority of, but not all, studies also support OCs being protective among carriers of BRCA/BRCA2 pathogenic variants. A meta-analysis of 18 studies including 13,627 carriers of BRCA pathogenic variants reported a significantly reduced risk of ovarian cancer (SRR, 0.50; 95% CI, 0.33–0.75) associated with OC use. (Refer to the Chemopreventive agents for reducing ovarian cancer risk section in BRCA1 and BRCA2: Cancer Risks and Management.)
Risk Factors for Endometrial Cancer
Refer to the PDQ summary on Endometrial Cancer Prevention for information about risk factors for endometrial cancer in the general population.
Age
Age is an important risk factor for endometrial cancer. Most women with endometrial cancer are diagnosed after menopause. Only 15% of women are diagnosed with endometrial cancer before age 50 years, and fewer than 5% are diagnosed before age 40 years. Women with Lynch syndrome tend to develop endometrial cancer at an earlier age, with the median age at diagnosis of 48 years.
Family history including inherited cancer genes
Although the hyperestrogenic state is the most common predisposing factor for endometrial cancer, family history also plays a significant role in a woman’s risk for disease. Approximately 3% to 5% of uterine cancer cases are attributable to a hereditary cause, with the main hereditary endometrial cancer syndrome being Lynch syndrome, an autosomal dominant genetic condition with a population prevalence of 1 in 300 to 1 in 1,000 individuals. (Refer to the Lynch Syndrome section in Genetics of Colorectal Cancer for more information.)
Non-Lynch syndrome genes may also contribute to endometrial cancer risk. In an unselected endometrial cancer cohort undergoing multigene panel testing, approximately 3% of patients tested positive for a germline pathogenic variant in non-Lynch syndrome genes, including CHEK2, APC, ATM, BARD1, BRCA1, BRCA2, BRIP1, NBN, PTEN, and RAD51C. Notably, patients with pathogenic variants in non-Lynch syndrome genes were more likely to have serous tumor histology than were patients without pathogenic variants. Furthermore, although the overall risk of endometrial cancer after RRSO was not increased among carriers of BRCA1 pathogenic variants, these patients seemed to have an increased risk of serous and serous-like endometrial cancer. These findings were supported by a Dutch multicenter cohort study in women with germline BRCA1 and BRCA2 pathogenic variants. This study concluded that participants' AR for endometrial cancer was approximately 3%. Because some serous and p53-aberrant endometrial cancers may harbor germline or somatic BRCA1/BRCA2 variants, poly (ADP-ribose) polymerase (PARP) inhibitor therapy may also be a therapeutic option.
Reproductive history
Reproductive factors such as multiparity, late menarche, and early menopause decrease the risk of endometrial cancer because of the lower cumulative exposure to estrogen and the higher relative exposure to progesterone.
Hormones
Hormonal factors that increase the risk of type I endometrial cancer are better understood. All endometrial cancers share a predominance of estrogen relative to progesterone. Prolonged exposure to estrogen or unopposed estrogen increases the risk of endometrial cancer. Endogenous exposure to estrogen can result from obesity, polycystic ovary syndrome, and nulliparity, while exogenous estrogen can result from taking unopposed estrogen or tamoxifen. Unopposed estrogen increases the risk of developing endometrial cancer by twofold to twentyfold, proportional to the duration of use. Tamoxifen, a selective estrogen receptor modulator, acts as an estrogen agonist on the endometrium while acting as an estrogen antagonist in breast tissue, and increases the risk of endometrial cancer. In contrast, OCs, the LNG-IUS, and combination estrogen-progesterone HRT all reduce the risk of endometrial cancer through the antiproliferative effect of progesterone acting on the endometrium.
Autosomal Dominant Inheritance of Breast and Gynecologic Cancer Predisposition
Autosomal dominant inheritance of breast and gynecologic cancers is characterized by transmission of cancer predisposition from generation to generation, through either the mother’s or the father’s side of the family, with the following characteristics:
- Inheritance risk of 50%. When a parent carries an autosomal dominant genetic predisposition, each child has a 50:50 chance of inheriting the predisposition. Although the risk of inheriting the predisposition is 50%, not everyone with the predisposition will develop cancer because of incomplete penetrance and/or gender-restricted or gender-related expression.
- Both males and females can inherit and transmit an autosomal dominant cancer predisposition. A male who inherits a cancer predisposition can still pass the altered gene on to his sons and daughters.
Breast and ovarian cancer are components of several autosomal dominant cancer syndromes. The syndromes most strongly associated with both cancers are the syndromes associated with BRCA1 or BRCA2 pathogenic variants. Breast cancer is also a common feature of Li-Fraumeni syndrome due to TP53 pathogenic variants and of PTEN hamartoma tumor syndromes (including Cowden syndrome) due to PTEN pathogenic variants. Other genetic syndromes that may include breast cancer as an associated feature include heterozygous carriers of the ATM gene and Peutz-Jeghers syndrome. Ovarian cancer has also been associated with Lynch syndrome, basal cell nevus (Gorlin) syndrome, and multiple endocrine neoplasia type 1. Lynch syndrome is mainly associated with colorectal cancer and endometrial cancer, although several studies have demonstrated that patients with Lynch syndrome are also at risk of developing transitional cell carcinoma of the ureters and renal pelvis; cancers of the stomach, small intestine, liver and biliary tract, brain, breast, prostate, and adrenal cortex; and sebaceous skin tumors (Muir-Torre syndrome).
Germline pathogenic variants in the genes responsible for these autosomal dominant cancer syndromes produce different clinical phenotypes of characteristic malignancies and, in some instances, associated nonmalignant abnormalities.
The family characteristics that suggest hereditary cancer predisposition include the following:
- Multiple cancers within a family.
- Cancers typically occur at an earlier age than in sporadic cases (defined as cases not associated with genetic risk).
- Two or more primary cancers in a single individual. These could be multiple primary cancers of the same type (e.g., bilateral breast cancer) or primary cancer of different types (e.g., breast cancer and ovarian cancer in the same individual or endometrial and colon cancer in the same individual).
- Cases of male breast cancer. The inheritance risk for autosomal dominant genetic conditions is 50% for both males and females, but the differing penetrance of the genes may result in some unaffected individuals in the family.
Figure 1 and Figure 2 depict some of the classic inheritance features of a BRCA1 and BRCA2 pathogenic variant, respectively. Figure 3 depicts a classic family with Lynch syndrome. For more information about pedigree nomenclature, see the Family history section in Cancer Genetics Risk Assessment and Counseling.
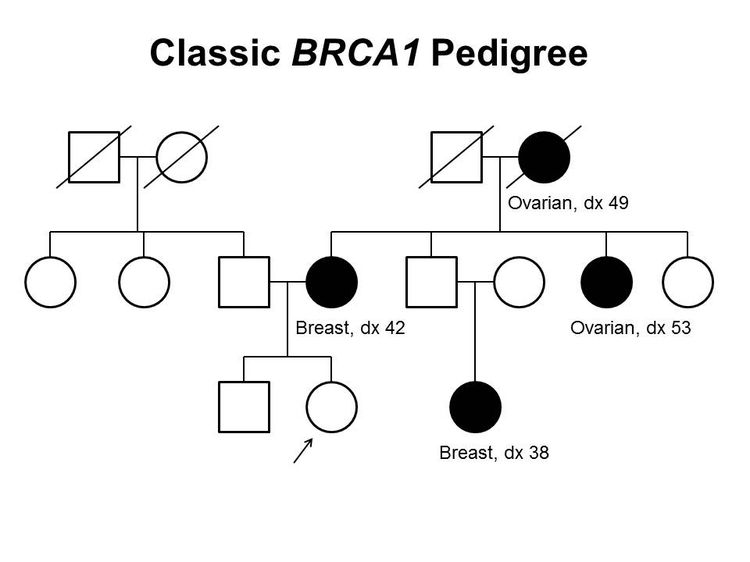 Figure 1. BRCA1 pedigree. This pedigree shows some of the classic features of a family with a BRCA1 pathogenic variant across three generations, including affected family members with breast cancer or ovarian cancer and a young age at onset. BRCA1 families may exhibit some or all of these features. As an autosomal dominant syndrome, a BRCA1 pathogenic variant can be transmitted through maternal or paternal lineages, as depicted in the figure.
Figure 1. BRCA1 pedigree. This pedigree shows some of the classic features of a family with a BRCA1 pathogenic variant across three generations, including affected family members with breast cancer or ovarian cancer and a young age at onset. BRCA1 families may exhibit some or all of these features. As an autosomal dominant syndrome, a BRCA1 pathogenic variant can be transmitted through maternal or paternal lineages, as depicted in the figure.
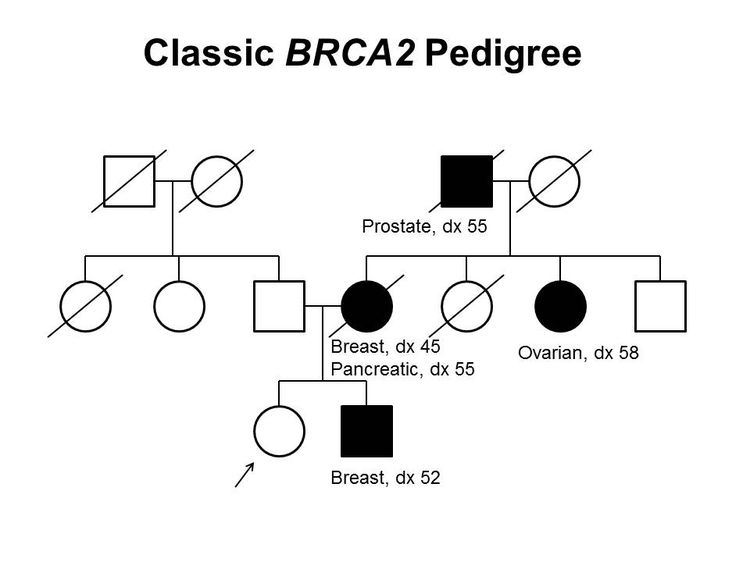 Figure 2. BRCA2 pedigree. This pedigree shows some of the classic features of a family with a BRCA2 pathogenic variant across three generations, including affected family members with breast (including male breast cancer), ovarian, pancreatic, or prostate cancers and a relatively young age at onset. BRCA2 families may exhibit some or all of these features. As an autosomal dominant syndrome, a BRCA2 pathogenic variant can be transmitted through maternal or paternal lineages, as depicted in the figure.
Figure 2. BRCA2 pedigree. This pedigree shows some of the classic features of a family with a BRCA2 pathogenic variant across three generations, including affected family members with breast (including male breast cancer), ovarian, pancreatic, or prostate cancers and a relatively young age at onset. BRCA2 families may exhibit some or all of these features. As an autosomal dominant syndrome, a BRCA2 pathogenic variant can be transmitted through maternal or paternal lineages, as depicted in the figure.
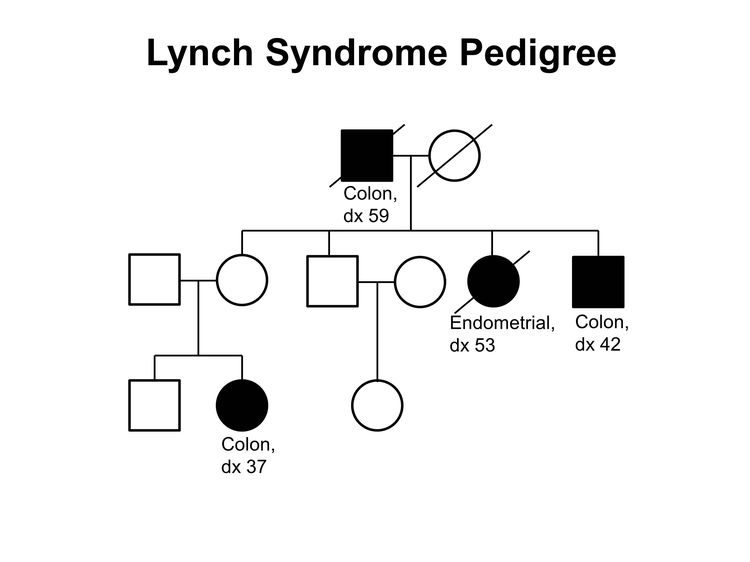 Figure 3. Lynch syndrome pedigree. This pedigree shows some of the classic features of a family with Lynch syndrome, including affected family members with colon cancer or endometrial cancer, a young age at onset in some individuals, and incomplete penetrance. Lynch syndrome families may exhibit some or all of these features. Lynch syndrome families may also include individuals with other gastrointestinal, gynecologic, and genitourinary cancers, or other extracolonic cancers. As an autosomal dominant syndrome, Lynch syndrome can be transmitted through maternal or paternal lineages, as depicted in the figure. Because the cancer risk is not 100%, individuals who have Lynch syndrome may not develop cancer, such as the mother of the female with colon cancer diagnosed at age 37 years in this pedigree (called incomplete penetrance).
Figure 3. Lynch syndrome pedigree. This pedigree shows some of the classic features of a family with Lynch syndrome, including affected family members with colon cancer or endometrial cancer, a young age at onset in some individuals, and incomplete penetrance. Lynch syndrome families may exhibit some or all of these features. Lynch syndrome families may also include individuals with other gastrointestinal, gynecologic, and genitourinary cancers, or other extracolonic cancers. As an autosomal dominant syndrome, Lynch syndrome can be transmitted through maternal or paternal lineages, as depicted in the figure. Because the cancer risk is not 100%, individuals who have Lynch syndrome may not develop cancer, such as the mother of the female with colon cancer diagnosed at age 37 years in this pedigree (called incomplete penetrance).
There are no pathognomonic features distinguishing breast and ovarian cancers occurring in carriers of BRCA1 or BRCA2 pathogenic variants from those occurring in noncarriers. Breast cancers occurring in carriers of BRCA1 pathogenic variants are more likely to be ER-negative, progesterone receptor (PR)–negative, human epidermal growth factor receptor two (HER2/neu)–negative (i.e., triple-negative breast cancers [TNBC]), and have a basal phenotype. BRCA1-associated ovarian cancers are more likely to be high-grade and of serous histopathology. (Refer to the BRCA1/2-associated breast cancer pathology and Pathologies of BRCA1/2-associated ovarian, fallopian tube, and primary peritoneal cancers sections in BRCA1 and BRCA2: Cancer Risks and Management for more information.)
Some pathologic features distinguish carriers of Lynch syndrome–associated pathogenic variants from noncarriers. The hallmark feature of endometrial cancers occurring in Lynch syndrome is mismatch repair (MMR) deficiencies, including the presence of microsatellite instability (MSI), and the absence of specific MMR proteins. In addition to these molecular changes, there are also histologic changes including tumor-infiltrating lymphocytes, peritumoral lymphocytes, undifferentiated tumor histology, lower uterine segment origin, and synchronous tumors.
Considerations in Risk Assessment and in Identifying a Family History of Breast and Ovarian Cancer Risk
The accuracy and completeness of family histories must be considered when they are used to assess risk. A reported family history may be erroneous, or a person may be unaware of relatives affected with cancer. In addition, small family sizes and premature deaths may limit the information obtained from a family history. Breast or ovarian cancer on the paternal side of the family usually involves more distant relatives than does breast or ovarian cancer on the maternal side, so information may be more difficult to obtain. When self-reported information is compared with independently verified cases, the sensitivity of a history of breast cancer is relatively high, at 83% to 97%, but lower for ovarian cancer, at 60%. Additional limitations of relying on family histories include adoption; families with a small number of women; limited access to family history information; and incidental removal of the uterus, ovaries, and/or fallopian tubes for noncancer indications. Family histories will evolve; therefore, it is important to update family histories from both parents over time. (Refer to the Accuracy of the family history section in Cancer Genetics Risk Assessment and Counseling for more information.)
Models for Prediction of Breast and Gynecologic Cancer Risk
Models to predict an individual’s lifetime risk of developing breast and/or gynecologic cancer are available. In addition, models exist to predict an individual’s likelihood of having a pathogenic variant in BRCA1, BRCA2, or one of the MMR genes associated with Lynch syndrome. (Refer to the Models for prediction of the likelihood of a BRCA1 or BRCA2 pathogenic variant section of this summary for more information about some of these models.) Not all models can be appropriately applied to all patients. Each model is appropriate only when the patient’s characteristics and family history are similar to those of the study population on which the model was based. Different models may provide widely varying risk estimates for the same clinical scenario, and the validation of these estimates has not been performed for many models.
Breast cancer risk assessment models
In general, breast cancer risk assessment models are designed for two types of populations: 1) women without a pathogenic variant or strong family history of breast or ovarian cancer; and 2) women at higher risk because of a personal or family history of breast cancer or ovarian cancer. Models designed for women of the first type (e.g., the Gail model, which is the basis for the Breast Cancer Risk Assessment Tool [BCRAT] , and the Colditz and Rosner model ) require only limited information about family history (e.g., number of FDRs with breast cancer). Models designed for women at higher risk require more detailed information about personal and family cancer history of breast and ovarian cancers, including ages at onset of cancer and/or carrier status of specific breast cancer-susceptibility alleles. The genetic factors used by the latter models differ, with some assuming one risk locus (e.g., the Claus model ), others assuming two loci (e.g., the International Breast Cancer Intervention Study [IBIS] model and the BRCAPRO model ), and still others assuming an additional polygenic component in addition to multiple loci (e.g., the Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm [BOADICEA] model ). The models also differ in whether they include information about nongenetic risk factors. Three models (Gail/BCRAT, Pfeiffer, and IBIS) include nongenetic risk factors but differ in the risk factors they include (e.g., the Pfeiffer model includes alcohol consumption, whereas the Gail/BCRAT does not). These models have limited ability to discriminate between individuals who are affected and those who are unaffected with cancer; a model with high discrimination would be close to 1, and a model with little discrimination would be close to 0.5; the discrimination of the models currently ranges between 0.56 and 0.63). The existing models generally are more accurate in prospective studies that have assessed how well they predict future cancers. An analysis comparing the 10-year performance of the BOADICEA, BRCAPRO, BCRAT, and IBIS models demonstrated superiority of the models with more detailed pedigree inclusion—specifically, BOADICEA and IBIS.
In the United States, BRCAPRO, the Claus model, and the Gail/BCRAT are widely used in clinical counseling. Risk estimates derived from the models differ for an individual patient. Several other models that include more detailed family history information are also in use and are discussed below.
Additional considerations for clinical use of breast cancer risk assessment models
The Gail model is the basis for the BCRAT, a computer program available from the National Cancer Institute by calling the Cancer Information Service at 1-800-4-CANCER (1-800-422-6237). This version of the Gail model estimates only the risk of invasive breast cancer. The Gail/BCRAT model has been found to be reasonably accurate at predicting breast cancer risk in large groups of White women who undergo annual screening mammography; however, reliability varies depending on the cohort studied. Risk can be overestimated in the following populations:
- Women who do not adhere to screening recommendations.
- Women in the highest-risk strata.
The Gail/BCRAT model is valid for women aged 35 years and older. The model was primarily developed for White women. Extensions of the Gail model for African American women have been subsequently developed to calibrate risk estimates using data from more than 1,600 African American women with invasive breast cancer and more than 1,600 controls. Additionally, extensions of the Gail model have incorporated high-risk single nucleotide variants (SNVs) and pathogenic variants; however, no software exists to calculate risk in these extended models. Other risk assessment models incorporating breast density have been developed but are not ready for clinical use.
Generally, the Gail/BCRAT model should not be the sole model used for families with one or more of the following characteristics:
- Multiple affected individuals with breast cancer or ovarian cancer (especially when one or more breast cancers are diagnosed before age 50 y).
- A woman with both breast and ovarian cancer.
- Ashkenazi Jewish ancestry with at least one case of breast or ovarian cancer (as these families are more likely to have a hereditary cancer susceptibility syndrome).
Commonly used models that incorporate family history include the IBIS, BOADICEA, and BRCAPRO models. The IBIS/Tyrer-Cuzick model incorporates both genetic and nongenetic factors. A three-generation pedigree is used to estimate the likelihood that an individual carries either a BRCA1/BRCA2 pathogenic variant or a hypothetical low-penetrance gene. In addition, the model incorporates personal risk factors such as parity, body mass index (BMI); height; and age at menarche, first live birth, menopause, and HRT use. Both genetic and nongenetic factors are combined to develop a risk estimate. The BOADICEA model examines family history to estimate breast cancer risk and also incorporates both BRCA1/BRCA2 and non-BRCA1/BRCA2 genetic risk factors. The most important difference between BOADICEA and the other models using information on BRCA1/BRCA2 is that BOADICEA assumes an additional polygenic component in addition to multiple loci, which is more in line with what is known about the underlying genetics of breast cancer. The BOADICEA model has also been expanded to include additional pathogenic variants, including CHEK2, ATM, and PALB2. However, the discrimination and calibration for these models differ significantly when compared in independent samples; the IBIS and BOADICEA models are more comparable when estimating risk over a shorter fixed time horizon (e.g., 10 years), than when estimating remaining lifetime risk. As all risk assessment models for cancers are typically validated over a shorter time horizon (e.g., 5 or 10 years), fixed time horizon estimates rather than remaining lifetime risk may be more accurate and useful measures to convey in a clinical setting.
In addition, readily available models that provide information about an individual woman’s risk in relation to the population-level risk depending on her risk factors may be useful in a clinical setting (e.g., Your Disease Risk). Although this tool was developed using information about average-risk women and does not calculate AR estimates, it still may be useful when counseling women about cancer prevention. Risk assessment models are being developed and validated in large cohorts to integrate genetic and nongenetic data, breast density, and other biomarkers.
Although most breast cancer risk models have been shown to be well calibrated overall, model performance can be different for subgroups of women. In particular, independent, prospective validation of risk models for women who tested negative for BRCA1 or BRCA2 pathogenic variants supported that the most commonly used clinical risk models underpredicted risk for this group of women. The performance also differed on the basis of whether the test results of relatives were known. The models also underpredicted risk by 26.3% to 56.7% in women who tested negative but whose relatives had not been tested.
Ovarian cancer risk assessment models
Two risk prediction models have been developed for ovarian cancer. The Rosner model included age at menopause, age at menarche, oral contraception use, and tubal ligation; the concordance statistic was 0.60 (0.57–0.62). The Pfeiffer model included oral contraceptive use, menopausal HRT use, and family history of breast cancer or ovarian cancer, with a similar discriminatory power of 0.59 (0.56–0.62). Although both models were well calibrated, their modest discriminatory power limited their screening potential.
Endometrial cancer risk assessment models
The Pfeiffer model has been used to predict endometrial cancer risk in the general population. For endometrial cancer, the RR model included BMI, menopausal HRT use, menopausal status, age at menopause, smoking status, and OC use. The discriminatory power of the model was 0.68 (0.66–0.70); it overestimated observed endometrial cancers in most subgroups but underestimated disease in women with the highest BMI category, in premenopausal women, and in women taking menopausal HRT for 10 years or more.
In contrast, MMRpredict, PREMM5 (PREdiction Model for gene Mutations), and MMRpro are three quantitative predictive models used to identify individuals who may potentially have Lynch syndrome. MMRpredict incorporates only colorectal cancer patients but does include MSI and immunohistochemistry (IHC) tumor testing results. PREMM5 is an update of PREMM (1,2,6) and includes each of the five genes associated with Lynch syndrome, including PMS2 and EPCAM. It accounts for other Lynch syndrome–associated tumors but does not include tumor testing results. MMRpro incorporates tumor testing and germline testing results, but is more time intensive because it includes affected and unaffected individuals in the risk-quantification process. All three predictive models are comparable to the traditional Amsterdam and Bethesda criteria in identifying individuals with colorectal cancer who carry MMR gene pathogenic variants. However, because these models were developed and validated in colorectal cancer patients, the discriminative abilities of these models to identify Lynch syndrome are lower among individuals with endometrial cancer than among those with colon cancer. In fact, the sensitivity and specificity of MSI and IHC in identifying carriers of pathogenic variants are considerably higher than the prediction models and support the use of molecular tumor testing to screen for Lynch syndrome in women with endometrial cancer.
Table 1 summarizes salient aspects of breast and gynecologic cancer risk assessment models that are commonly used in the clinical setting. These models differ by the extent of family history included, whether nongenetic risk factors are included, and whether carrier status and polygenic risk are included (inputs to the models). The models also differ in the type of risk estimates that are generated (outputs of the models). These factors may be relevant in choosing the model that best applies to a particular individual.
| Model | Family History (input) | Pathogenic Variants (input) | Risk Factors (input) | Risk Estimate Generated (output) |
|---|---|---|---|---|
| BCRAT = Breast Cancer Risk Assessment Tool; BOADICEA = Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm; IBIS = International Breast Cancer Intervention Study; PREMM = PREdiction Model for gene Mutations. | ||||
| aHigh risk is defined as those with a personal or family history of the designated cancer type. | ||||
| bTakes into account polygenes as an underlying assumption of the model. | ||||
| Breast Cancer Risk Assessment Models | ||||
| Models for Average-Risk Women | ||||
| Gail/BCRAT | First-degree relatives (breast cancer) | No | Yes | Breast cancer |
| Pfeiffer (breast) | First-degree relatives (breast, ovarian cancers) | No | Yes | Breast cancer |
| Colditz and Rosner | None | No | Yes | Breast cancer |
| Models for High-Risk Womena | ||||
| Claus | Multigenerational (breast cancer) | No | No | Breast cancer |
| BRCAPRO | Multigenerational (breast, ovarian cancers) | BRCA1/BRCA2 | No | Breast cancer; % risk of carrying BRCA1/BRCA2 pathogenic variant |
| IBIS | Multigenerational (ovarian cancer) | BRCA1/BRCA2 | Yes | Breast cancer; % risk of carrying BRCA1/BRCA2 pathogenic variant |
| BOADICEA (CanRisk tool)b | Multigenerational (pancreatic, breast, ovarian cancers) | BRCA1/BRCA2 | No | Breast and ovarian cancer; % risk of carrying BRCA1/BRCA2 pathogenic variant |
| Ovarian Cancer Risk Assessment Models | ||||
| Models for Average-Risk Women | ||||
| Rosner | None | No | Yes | Ovarian cancer |
| Pfeiffer (ovarian) | First-degree relatives (breast, ovarian cancers) | No | Yes | Breast cancer |
| Models for High-Risk Womena | ||||
| BOADICEA (CanRisk tool)b | Multigenerational (pancreatic, breast, ovarian cancers) | BRCA1/BRCA2 | No | Breast and ovarian cancer; % risk of carrying BRCA1/BRCA2 pathogenic variant |
| Endometrial Cancer Risk Assessment Models | ||||
| Models for Average-Risk Women | ||||
| Pfeiffer (endometrial) | None | No | Yes | Endometrial cancer |
| Models for High-Risk Womena | ||||
| PREMM5 | Multigenerational (colon, endometrial and other Lynch syndrome–associated cancers and polyps) | No | No | % risk of carrying MLH1, MSH2, MSH6 pathogenic variant |
| MMRpro | Multigenerational (colon, endometrial cancers) | No | No | % risk of carrying MLH1, MSH2, MSH6 pathogenic variant |
| MMRpredict | Multigenerational (colon, endometrial cancers) | No | No | % risk of carrying MLH1, MSH2, MSH6 pathogenic variant |
Models for Predicting the Likelihood of a BRCA1/BRCA2 Pathogenic Variant
Many models have been developed to predict the probability of identifying germline BRCA1/BRCA2 pathogenic variants in individuals or families. These models include those using logistic regression, genetic models using Bayesian analysis (BRCAPRO and BOADICEA), and empiric observations.
In addition to BOADICEA, BRCAPRO is commonly used for genetic counseling in the clinical setting. BRCAPRO and BOADICEA predict the probability of being a carrier and produce estimates of breast cancer risk (refer to Table 2). The discrimination and accuracy (factors used to evaluate the performance of prediction models) of these models are much higher for their ability to report on carrier status than for their ability to predict fixed or remaining lifetime risk.
BOADICEA is a polygenetic model that uses complex segregation analysis to examine both breast cancer risk and the probability of having a BRCA1 or BRCA2 pathogenic variant. Even among experienced providers, the use of prediction models has been shown to increase the power to discriminate which patients are most likely to be carriers of BRCA1/BRCA2 pathogenic variants. Most models do not include other cancers seen in the BRCA1 and BRCA2 spectrum, such as pancreatic cancer and prostate cancer. Interventions that decrease the likelihood that an individual will develop cancer (such as oophorectomy and mastectomy) may influence the ability to predict BRCA1 and BRCA2 pathogenic variant status. One study has shown that the prediction models for genetic risk are sensitive to the amount of family history data available and do not perform as well with limited family information. BOADICEA is being expanded to incorporate additional risk variants (genome-wide association studies [GWAS] and SNVs) to better predict pathogenic variant status and to improve the accuracy of breast cancer and ovarian cancer risk estimates.
The performance of the models can vary in specific ethnic groups. The BRCAPRO model appeared to best fit a series of French Canadian families. There have been variable results in the performance of the BRCAPRO model among Hispanic individuals, and both the BRCAPRO model and Myriad tables underestimated the proportion of carriers of pathogenic variants in an Asian American population. BOADICEA was developed and validated in British women. Thus, the major models used for both overall risk (Table 1) and genetic risk (Table 2) have not been developed or validated in large populations of racially and ethnically diverse women. Of the commonly used clinical models for assessing genetic risk, only the Tyrer-Cuzick model contains nongenetic risk factors.
The power of several of the models has been compared in different studies. Four breast cancer genetic-risk models, BOADICEA, BRCAPRO, IBIS, and eCLAUS, were evaluated for their diagnostic accuracy in predicting BRCA1/BRCA2 pathogenic variants in a cohort of 7,352 German families. The family member with the highest likelihood of carrying a pathogenic variant from each family was screened for BRCA1/BRCA2 pathogenic variants. Carrier probabilities from each model were calculated and compared with the actual variants detected. BRCAPRO and BOADICEA had significantly higher diagnostic accuracy than IBIS or eCLAUS. Accuracy for the BOADICEA model was further improved when statuses of the tumor markers ER, PR, and HER2/neu were included in the model. The inclusion of these biomarkers has been shown to improve the performance of BRCAPRO.
| Myriad Prevalence Tables | BRCAPRO | BOADICEA | Tyrer-Cuzick | |
|---|---|---|---|---|
| AJ = Ashkenazi Jewish; BOADICEA = Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm; FDR = first-degree relatives; SDR = second-degree relatives. | ||||
| Method | Empiric data from Myriad Genetics based on personal and family history reported on requisition forms | Statistical model, assumes autosomal dominant inheritance | Statistical model, assumes polygenic risk | Statistical model, assumes autosomal dominant inheritance |
| Features of the model | Proband may or may not have breast or ovarian cancer | Proband may or may not have breast or ovarian cancer | Proband may or may not have breast or ovarian cancer | Proband must be unaffected |
| Considers age of breast cancer diagnosis as <50 y, >50 y | Considers exact age at breast and ovarian cancer diagnosis | Considers exact age at breast and ovarian cancer diagnosis | Also includes reproductive factors and body mass index to estimate breast cancer risk | |
| Considers breast cancer in ≥1 affected relative only if diagnosed <50 y | Considers prior genetic testing in family (i.e., BRCA1/BRCA2 pathogenic variant–negative relatives) | Includes all FDR and SDR with and without cancer | ||
| Considers ovarian cancer in ≥1 relative at any age | Considers oophorectomy status | Includes AJ ancestry | ||
| Includes AJ ancestry | Includes all FDR and SDR with and without cancer | |||
| Very easy to use | Includes AJ ancestry | |||
| Limitations | Simplified/limited consideration of family structure | Requires computer software and time-consuming data entry | Requires computer software and time-consuming data entry | Designed for individuals unaffected with breast cancer |
| Incorporates only FDR and SDR; may need to change proband to best capture risk and to account for disease in the paternal lineage | ||||
| May overestimate risk in bilateral breast cancer | ||||
| Early age of breast cancer onset | May perform better in White populations than in racial and ethnic minority populations | Incorporates only FDR and SDR; may need to change proband to best capture risk | ||
| May underestimate risk of BRCA pathogenic variant in high-grade serous ovarian cancers but overestimate the risk for other histologies | ||||
Genetic testing for BRCA1 and BRCA2 pathogenic variants has been available to the public since 1996. As more individuals have undergone testing, risk assessment models have improved. This, in turn, gives providers better data to estimate an individual patient’s risk of carrying a pathogenic variant, but risk assessment continues to be an art. There are factors that might limit the ability to provide an accurate risk assessment (i.e., small family size, paucity of women, or ethnicity) including the specific circumstances of the individual patient (such as history of disease or risk-reducing surgeries).
Considerations When Conducting Genetic Testing
Indications for hereditary breast and gynecologic cancers genetic testing
Several professional organizations and expert panels—including the American Society of Clinical Oncology, the National Comprehensive Cancer Network (NCCN), the American Society of Human Genetics, the American College of Medical Genetics and Genomics, the National Society of Genetic Counselors, the U.S. Preventive Services Task Force, and the Society of Gynecologic Oncologists —have developed clinical criteria and practice guidelines that can be helpful to health care providers in identifying individuals who may have a BRCA1 or BRCA2 pathogenic variant.
In 2019, the American Society of Breast Surgeons published a recommendation to make genetic testing for “BRCA1/BRCA2, and PALB2, with other genes as appropriate for the clinical scenario and family history” available to all breast cancer patients. This recommendation was based on a study that suggested similar pathogenic variant rates identified through an extended multigene panel in patients with breast cancer who did or did not meet the NCCN guidelines for genetic testing. This study had important methodologic challenges that need to be considered, including exclusion of participants previously tested, uncertain accuracy of the reported risk criteria for study participants, inclusion of genes with uncertain management guidelines, and difference in the specific genes in which pathogenic or likely pathogenic variants were identified across the two groups. For example, there was a statistically significant difference between participants who met and who did not meet NCCN criteria in the detection of BRCA1/BRCA2 variants.
Other studies have also found that the NCCN criteria have good sensitivity when predicting BRCA1/BRCA2 variants; however, less is known about many other genes. For example, one study showed that the NCCN criteria were able to detect 88.9% of the BRCA1/BRCA2 pathogenic variant carriers and others have found that, if more than one NCCN criterion is met, then the positive predictive value does pass the 10% threshold (e.g., 12% for more than two NCCN criteria).
As the cost of genetic testing continues to decrease, there is a need for unbiased evidence to guide indications for testing, including the cost-benefit impact on screening, prevention, and treatment. Efforts to generate less biased evidence include a single institution study of 3,907 unselected women with breast cancer tested for nine breast cancer genes, including BRCA1/BRCA2, ATM, CDH1, CHEK2, NF1, PALB2, PTEN, and TP53. The study assessed the relative performance of NCCN genetic testing criteria as compared with the American Society of Breast Surgeons' recommendation to test all women aged 65 years or younger with breast cancer. The sensitivity of the criteria was defined as the proportion of individuals who met testing criteria and tested positive for a pathogenic or likely pathogenic variant of the total population of pathogenic or likely pathogenic variant carriers in the study, while the specificity was defined as the proportion of individuals who did not meet testing criteria and tested negative for a pathogenic or likely pathogenic variant of the total population of noncarriers in the study. High sensitivity and specificity are both important considerations; however, higher sensitivity leads to lower specificity, so it is important to balance these two factors. Detection of BRCA1/BRCA2 pathogenic or likely pathogenic variants based on NCCN criteria had a sensitivity of 87% with a specificity of 53.5%; when expanded to the nine genes included in the study, sensitivity was 70% and specificity was 53.2%. When including all women diagnosed with breast cancer at age 65 or younger, the sensitivity to detect BRCA1/BRCA2 pathogenic or likely pathogenic variants increased to 98%, while the specificity dropped to 22%. Among those who did not meet NCCN criteria, 0.7% had pathogenic or likely pathogenic BRCA1/BRCA2 variants.
Another study to assess frequency of pathogenic or likely pathogenic variants among breast cancer patients included a nested case-control study conducted through the WHI cohort among women with (cases) and without (controls) invasive breast cancer. Participants were tested for pathogenic or likely pathogenic variants in ten breast cancer–associated genes, including BRCA1/BRCA2. The prevalence of pathogenic or likely pathogenic BRCA1/BRCA2 variants among those diagnosed with invasive breast cancer before age 65 years was 2.21%, compared with 1.09% among those diagnosed at age 65 years or older. In comparison, the frequency of pathogenic or likely pathogenic BRCA1/BRCA2 variants was 0.22% in the control group. Current genetic testing criteria detect BRCA pathogenic variants. Although higher sensitivity is always desired, it is at the expense of specificity. Lower specificity leads to higher costs to achieve one positive genetic test.
Benefits of offering genetic testing at the time of cancer diagnosis
At the time of a new cancer diagnosis, genetic testing for inherited cancer predisposition may guide patient care including decisions about surgery, chemotherapy and other biologics, and radiation treatment. Among high-risk patients, the option of genetic testing is an important part of the shared decision-making process regarding cancer treatments at the time of diagnosis. Tools are available to facilitate decision making about genetic testing in this context.
Breast cancer diagnosis
Benefits of offering genetic testing at the time of breast cancer diagnosis include, but are not limited to, the following:
- Surgery: The identification of inherited susceptibility to breast cancer may influence surgical treatment decisions. As an example, the high risk of a second primary breast cancer among BRCA pathogenic variant carriers, particularly those diagnosed at an early age, may influence their decision to choose a bilateral mastectomy (versus a lumpectomy or unilateral/subtotal mastectomy) for surgical treatment of their breast cancer. Discussion of RRSO is indicated, and referral to a gynecologic provider may be considered.
- Chemotherapy and other biologics: Medical treatments may be guided by the identification of a pathogenic variant in an inherited cancer predisposing gene. As an example, among BRCA pathogenic variant carriers, breast cancer treatment may include the use of platinum-based agents. Furthermore, novel agents such as PARP inhibitors may be used in the treatment of metastatic breast cancer.
- Radiation therapy: Decisions about the use of radiation treatment may be guided by the presence of a pathogenic variant in an inherited breast cancer susceptibility gene. In particular, the poorer wound healing in irradiated breasts is an important consideration for those who may consider risk-reducing mastectomy with reconstruction. As an example, individuals with a pathogenic variant in TP53 may experience higher risks from radiation, including increased risks for subsequent new cancers. Thus, identification of TP53 carriers in the context of an active breast cancer diagnosis may influence radiation treatment decisions and reconstruction options.
Ovarian cancer diagnosis
Benefits of offering genetic testing at the time of ovarian cancer diagnosis include, but are not limited to, the following:
- Surgery: In most cases, the decision for ovarian cancer surgery is made on the basis of an adnexal mass or abdominal symptoms. When possible, considering the likelihood of a heritable genetic variant at the time of diagnosis may add value to surgical decision-making. The identification of inherited susceptibility to ovarian/fallopian tube cancer may influence surgical treatment decisions. For a questionable adnexal mass in a younger woman who is at risk of carrying a pathogenic variant of a highly penetrant ovarian cancer gene, knowledge of this information may help guide a decision for risk-reducing or therapeutic surgery. For women who may be considering fertility preservation surgery, genetic knowledge may motivate consideration of bilateral salpingo-oophorectomy, and in the case of carriers of BRCA1 pathogenic variants, a more detailed discussion regarding aggressive uterine cancer risk.
- Chemotherapy and other biologics: First-line chemotherapy for ovarian cancer still relies on a backbone of platinum and taxane chemotherapy. Current treatment options for optimally resected stage III ovarian carcinoma include intravenous (IV) chemotherapy, dose-dense IV chemotherapy, and a combination of IV paclitaxel plus intraperitoneal (IP) cisplatin, followed by IP paclitaxel 1 week later. Carriers of BRCA1 and BRCA2 pathogenic variants are considered more platinum sensitive, with longer progression-free survival times compared with BRCA1 and BRCA2 wild-type patients, so it is unclear whether a particular treatment strategy is driven more by antiangiogenesis effects, peritoneal dose intensity, or platinum dose intensity. The advent of PARP as a biologic target (in combination with chemotherapy or as maintenance) may also increase the armory of first-line treatment of ovarian cancer. (Refer to the Ovarian Cancer Treatment Strategies section in BRCA1 and BRCA2: Cancer Risks and Management for more information about PARP inhibitors in ovarian cancer treatment.)
Endometrial cancer diagnosis
Benefits of offering genetic testing at the time of endometrial cancer diagnosis include, but are not limited to, the following:
- Surgery: The most common treatment for a newly diagnosed endometrial cancer includes hysterectomy with removal of the ovaries and fallopian tubes, as well as assessment of lymph nodes. An exception to this practice might apply to a younger woman who wishes to retain fertility or retain her adnexa. IHC of endometrial sampling may allow for an assessment of the likelihood of a heritable genetic variant at the time of diagnosis, which may add value to the surgical decision-making process. For a young woman who is found to have Lynch syndrome, knowledge of this information may help guide a decision for hormonal management of endometrial cancer to allow future childbearing, or RRSO if her risk of ovarian cancer is deemed high enough on the basis of a specific genetic variant. For a young woman who is found to carry a pathogenic variant in BRCA1/BRCA2, or one of the other homologous recombination deficiencies increasing ovarian cancer risk, she may wish to decide between salpingo-oophorectomy or, at least, salpingectomy.
- Chemotherapy and other biologics: Immune checkpoint inhibitors are now approved for use in endometrial cancers that have MSI or MMR deficiency. While MSI and MMR status can be assessed at either the time of diagnosis or recurrent disease, it may be beneficial to perform tumor testing at diagnosis with the primary pathology processing, usually at the time of hysterectomy.
Multigene (panel) testing
Since the availability of next-generation sequencing and the Supreme Court of the United States ruling that human genes cannot be patented, several clinical laboratories now offer genetic testing through multigene panels at a cost comparable to that of single-gene testing. Even testing for BRCA1 and BRCA2 is a limited panel test of two genes. Approximately 25% of all ovarian/fallopian tube/peritoneal cancers are caused by a heritable genetic condition. Of these, about one-quarter (6% of all ovarian/fallopian tube/peritoneal cancers) are caused by genes other than BRCA1 and BRCA2, including many genes associated with the Fanconi anemia pathway or otherwise involved with homologous recombination. In a population of ovarian cancer patients who test negative for BRCA1 and BRCA2 pathogenic variants, multigene panel testing can reveal actionable pathogenic variants.
In general, multigene panel testing increases the yield of non-BRCA pathogenic variants across a variety of populations. In an unselected population of breast cancer patients, the prevalence of BRCA1 and BRCA2 pathogenic variants was 6.1%, while the prevalence of pathogenic variants in other breast/ovarian cancer–predisposing genes was 4.6%. In an unselected population of endometrial cancer patients, the prevalence of Lynch syndrome pathogenic variants (MLH1, MSH2, EPCAM-MSH2, MSH6, and PMS2) was 5.8%; the prevalence of pathogenic variants in other actionable genes was 3.4%. Similarly, in a study of 35,409 women with breast cancer tested with the Myriad 25-gene panel, a pathogenic variant was found in 9.3% of women. Among that 9.3%, 48.5% of the women carried a pathogenic variant in BRCA1 or BRCA2. The majority of other breast cancer genes with pathogenic variants identified included CHEK2 (11.7%), ATM (9.7%), and PALB2 (9.3%). The prevalence of pathogenic variants in the other breast cancer genes on the panel ranged from 0.05% to 0.31%. Pathogenic variants in Lynch syndrome genes accounted for 7.0% of variants identified; 3.7% were found in other genes included in the panel. The rate of pathogenic variants was higher in women with TNBC diagnosed before age 40 years. A similar trend of identifying pathogenic variants in non-BRCA susceptibility genes in male breast cancer patients has also been described. In two studies of women who had previously tested negative for BRCA1/BRCA2, reflex testing with a multigene panel identified pathogenic variants in additional genes among 8% to 11% of cases. In a study of 77,085 patients with breast cancer and 6,001 patients with ovarian cancer, 24.1% and 30.9% had genetic testing, respectively. Of those tested, pathogenic or likely pathogenic variants were identified in 7.8% of patients with breast cancer and 14.5% of patients with ovarian cancer. Prevalent non-BRCA pathogenic variants identified in patients with breast cancer included CHEK2 (1.6%), PALB2 (1.0%), ATM (0.7%), and NBN (0.4%). In patients with ovarian cancer, non-BRCA pathogenic variants included CHEK2 (1.4%), BRIP1 (0.9%), MSH2 (0.8%), and ATM (0.6%). The potential utility of genetic testing in patients with ovarian tumors of all histologies was suggested in a study using a 32-gene panel that found 13.2% of 4,439 tumors harbored a pathogenic variant. Rates were highest among those with serous ovarian carcinoma (14.7%), although likely pathogenic variants were also seen in those with other histologies (borderline, germ cell, and sex cord stromal tumors), the significance of which is unclear to clinical management or etiology of disease.
Multi-gene panel testing was conducted as part of two large efforts led by the worldwide Breast Cancer Association Consortium (BCAC) and the United States–based CARRIERS consortium. The BCAC study tested 113,927 women for 34 inherited cancer genes, while the CARRIERS study tested 64,791 women for 28 hereditary cancer genes. In both studies, significant associations were reported between eight genes and breast cancer development (BRCA1, BRCA2, PALB2, BARD1, RAD51C, RAD51D, ATM, and CHEK2). Associations were only reported between MSH6 and breast cancer development in the BCAC study. Similarly, associations were only reported between CDH1 and breast cancer development in the CARRIERS study. Both TP53 and PTEN (which are established breast cancer risk genes that are linked to early-onset disease) were not significantly associated with breast cancer development in these studies. This is presumably because TP53 and PTEN pathogenic variants are very rare.
NCCN recommends that women diagnosed with TNBC undergo BRCA1/BRCA2, CDH1, PALB2, PTEN, STK11, and TP53 testing to guide treatment decisions at any age. A large study utilizing multigene (panel) testing comprising two separate cohorts reported that, in addition to BRCA1/BRCA2 genes, six other breast cancer susceptibility genes were also related to a higher risk of TNBC. Specifically, pathogenic variants in BARD1, PALB2, and RAD51D, in addition to BRCA1 and BRCA2, were each associated with more than a fivefold increase in breast cancer. Pathogenic variants in three other genes —BRIP1, RAD51C, and TP53— were each associated with an increased TNBC risk of more than twofold. Pathogenic variants in these eight genes were reported in 12% of the TNBC cases (8.3% BRCA1/BRCA2, 3.7% non-BRCA1/BRCA2). The study was conducted in a clinical testing cohort of 140,449 individuals (8,753 TNBC cases) who received genetic testing using a 21-gene panel (sample A). In addition, a second sample (sample B) examined gene frequency rates in a pooled consortium of 2,143 individuals using a 17-gene panel. The overall frequency of pathogenic variants in the 21 genes examined in sample A was 14.4% (8.4% BRCA1/BRCA2, 6.0% non-BRCA1/BRCA2). The two samples had very consistent findings with respect to the risk estimates despite differences in age, race, ethnicity, and family history of cancer with sample A being younger, more racially and ethnically diverse, and more likely to have a family history of cancer. The pathogenic variant frequency detection in these 21 genes was also similar for White individuals (14% overall, 7.8% BRCA1/BRCA2, 6.2% non-BRCA1/BRCA2) and African American individuals (14.6% overall, 9.0% BRCA1/BRCA2, 5.6% non-BRCA1/BRCA2).
Multi-gene panel testing studies were conducted in women from the United States who had African ancestry, and results showed that certain genes were associated with increased breast cancer risk in this population. These genes were similar to the breast cancer risk genes found in individuals from the United States with European ancestry. A case-control study of 10,047 women with African ancestry found a pathogenic variant frequency of 10.3% in those with ER-negative breast cancer, 5.2% in those with ER-positive breast cancer, and 2.3% in those without breast cancer. BRCA1 (OR, 47), BRCA2 (OR, 7.25) and PALB2 (OR, 8.54) were associated with the highest breast cancer risks. High ER-negative breast cancer risk was reported in individuals with pathogenic variants in RAD51D (OR, 7.82), while moderate ER-positive breast cancer risk was reported in individuals with pathogenic variants in CHEK2, ATM, ERCC3, and FANCC. Similarly, a case-control study of 3,286 women with African ancestry found significant associations between breast cancer risk and pathogenic variants in the following genes: BRCA1, BRCA2, PALB2, ATM, CHEK2, TP53, NF1, RAD51C, and RAD51D.
There are caveats of multigene testing. Genes identified as part of multigene panel testing can be associated with varied breast cancer risk or confer no known risk. There is also the possibility of finding a variant of uncertain significance (VUS). Even within a given gene, there may be differential risks on the basis of specific pathogenic variants. A large population-based retrospective study using Surveillance, Epidemiology, and End Results (SEER) program data from Georgia and Los Angeles, California, found that multigene testing led to a twofold increase in the detection of pathogenic variants compared with BRCA-only testing in women with breast cancer. VUS rates, however, were tenfold higher in the multigene panels, especially in African American women (44.5%) and Asian women (50.9%). Many centers now offer a multigene panel test instead of just BRCA1 and BRCA2 testing if there is a concerning family history of syndromes other than hereditary breast and ovarian cancer, or more importantly, to gain as much genetic information as possible with one test, particularly if there may be insurance limitations.
(Refer to the Multigene [panel] testing section in Cancer Genetics Risk Assessment and Counseling for more information about multigene testing, including genetic education and counseling considerations and research examining the use of multigene testing.)
References
- American Cancer Society: Cancer Facts and Figures 2024. American Cancer Society, 2024. Available online. Last accessed January 17, 2024.
- Ravdin PM, Cronin KA, Howlader N, et al.: The decrease in breast-cancer incidence in 2003 in the United States. N Engl J Med 356 (16): 1670-4, 2007.
- Quante AS, Herz J, Whittemore AS, et al.: Assessing absolute changes in breast cancer risk due to modifiable risk factors. Breast Cancer Res Treat 152 (1): 193-7, 2015.
- Feuer EJ, Wun LM, Boring CC, et al.: The lifetime risk of developing breast cancer. J Natl Cancer Inst 85 (11): 892-7, 1993.
- Yang Q, Khoury MJ, Rodriguez C, et al.: Family history score as a predictor of breast cancer mortality: prospective data from the Cancer Prevention Study II, United States, 1982-1991. Am J Epidemiol 147 (7): 652-9, 1998.
- Colditz GA, Willett WC, Hunter DJ, et al.: Family history, age, and risk of breast cancer. Prospective data from the Nurses' Health Study. JAMA 270 (3): 338-43, 1993.
- Slattery ML, Kerber RA: A comprehensive evaluation of family history and breast cancer risk. The Utah Population Database. JAMA 270 (13): 1563-8, 1993.
- Johnson N, Lancaster T, Fuller A, et al.: The prevalence of a family history of cancer in general practice. Fam Pract 12 (3): 287-9, 1995.
- Pharoah PD, Day NE, Duffy S, et al.: Family history and the risk of breast cancer: a systematic review and meta-analysis. Int J Cancer 71 (5): 800-9, 1997.
- Bevier M, Sundquist K, Hemminki K: Risk of breast cancer in families of multiple affected women and men. Breast Cancer Res Treat 132 (2): 723-8, 2012.
- Kharazmi E, Chen T, Narod S, et al.: Effect of multiplicity, laterality, and age at onset of breast cancer on familial risk of breast cancer: a nationwide prospective cohort study. Breast Cancer Res Treat 144 (1): 185-92, 2014.
- Reiner AS, Sisti J, John EM, et al.: Breast Cancer Family History and Contralateral Breast Cancer Risk in Young Women: An Update From the Women's Environmental Cancer and Radiation Epidemiology Study. J Clin Oncol 36 (15): 1513-1520, 2018.
- Albright FS, Kohlmann W, Neumayer L, et al.: Population-based relative risks for specific family history constellations of breast cancer. Cancer Causes Control 30 (6): 581-590, 2019.
- Mucci LA, Hjelmborg JB, Harris JR, et al.: Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 315 (1): 68-76, 2016.
- Chen J, Pee D, Ayyagari R, et al.: Projecting absolute invasive breast cancer risk in white women with a model that includes mammographic density. J Natl Cancer Inst 98 (17): 1215-26, 2006.
- Dupont WD, Page DL, Parl FF, et al.: Long-term risk of breast cancer in women with fibroadenoma. N Engl J Med 331 (1): 10-5, 1994.
- Zeinomar N, Phillips KA, Daly MB, et al.: Benign breast disease increases breast cancer risk independent of underlying familial risk profile: Findings from a Prospective Family Study Cohort. Int J Cancer 145 (2): 370-379, 2019.
- Boyd NF, Byng JW, Jong RA, et al.: Quantitative classification of mammographic densities and breast cancer risk: results from the Canadian National Breast Screening Study. J Natl Cancer Inst 87 (9): 670-5, 1995.
- Byrne C, Schairer C, Wolfe J, et al.: Mammographic features and breast cancer risk: effects with time, age, and menopause status. J Natl Cancer Inst 87 (21): 1622-9, 1995.
- Pankow JS, Vachon CM, Kuni CC, et al.: Genetic analysis of mammographic breast density in adult women: evidence of a gene effect. J Natl Cancer Inst 89 (8): 549-56, 1997.
- Boyd NF, Lockwood GA, Martin LJ, et al.: Mammographic densities and risk of breast cancer among subjects with a family history of this disease. J Natl Cancer Inst 91 (16): 1404-8, 1999.
- Vachon CM, King RA, Atwood LD, et al.: Preliminary sibpair linkage analysis of percent mammographic density. J Natl Cancer Inst 91 (20): 1778-9, 1999.
- Vachon CM, van Gils CH, Sellers TA, et al.: Mammographic density, breast cancer risk and risk prediction. Breast Cancer Res 9 (6): 217, 2007.
- Mitchell G, Antoniou AC, Warren R, et al.: Mammographic density and breast cancer risk in BRCA1 and BRCA2 mutation carriers. Cancer Res 66 (3): 1866-72, 2006.
- Ramón Y Cajal T, Chirivella I, Miranda J, et al.: Mammographic density and breast cancer in women from high risk families. Breast Cancer Res 17: 93, 2015.
- Thompson CM, Mallawaarachchi I, Dwivedi DK, et al.: The Association of Background Parenchymal Enhancement at Breast MRI with Breast Cancer: A Systematic Review and Meta-Analysis. Radiology 292 (3): 552-561, 2019.
- Terry MB, Liao Y, Kast K, et al.: The Influence of Number and Timing of Pregnancies on Breast Cancer Risk for Women With BRCA1 or BRCA2 Mutations. JNCI Cancer Spectr 2 (4): pky078, 2018.
- Johannsson O, Loman N, Borg A, et al.: Pregnancy-associated breast cancer in BRCA1 and BRCA2 germline mutation carriers. Lancet 352 (9137): 1359-60, 1998.
- Jernström H, Lerman C, Ghadirian P, et al.: Pregnancy and risk of early breast cancer in carriers of BRCA1 and BRCA2. Lancet 354 (9193): 1846-50, 1999.
- Friebel TM, Domchek SM, Rebbeck TR: Modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: systematic review and meta-analysis. J Natl Cancer Inst 106 (6): dju091, 2014.
- Valentini A, Lubinski J, Byrski T, et al.: The impact of pregnancy on breast cancer survival in women who carry a BRCA1 or BRCA2 mutation. Breast Cancer Res Treat 142 (1): 177-85, 2013.
- Jernström H, Lubinski J, Lynch HT, et al.: Breast-feeding and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst 96 (14): 1094-8, 2004.
- Breast cancer and hormonal contraceptives: collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Collaborative Group on Hormonal Factors in Breast Cancer. Lancet 347 (9017): 1713-27, 1996.
- Iodice S, Barile M, Rotmensz N, et al.: Oral contraceptive use and breast or ovarian cancer risk in BRCA1/2 carriers: a meta-analysis. Eur J Cancer 46 (12): 2275-84, 2010.
- Schrijver LH, Olsson H, Phillips KA, et al.: Oral Contraceptive Use and Breast Cancer Risk: Retrospective and Prospective Analyses From a BRCA1 and BRCA2 Mutation Carrier Cohort Study. JNCI Cancer Spectr 2 (2): pky023, 2018.
- Huber D, Seitz S, Kast K, et al.: Use of oral contraceptives in BRCA mutation carriers and risk for ovarian and breast cancer: a systematic review. Arch Gynecol Obstet 301 (4): 875-884, 2020.
- Kotsopoulos J, Lubinski J, Moller P, et al.: Timing of oral contraceptive use and the risk of breast cancer in BRCA1 mutation carriers. Breast Cancer Res Treat 143 (3): 579-86, 2014.
- Conz L, Mota BS, Bahamondes L, et al.: Levonorgestrel-releasing intrauterine system and breast cancer risk: A systematic review and meta-analysis. Acta Obstet Gynecol Scand 99 (8): 970-982, 2020.
- Breast cancer and hormone replacement therapy: collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Collaborative Group on Hormonal Factors in Breast Cancer. Lancet 350 (9084): 1047-59, 1997.
- Gorsky RD, Koplan JP, Peterson HB, et al.: Relative risks and benefits of long-term estrogen replacement therapy: a decision analysis. Obstet Gynecol 83 (2): 161-6, 1994.
- Writing Group for the Women's Health Initiative Investigators: Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women's Health Initiative randomized controlled trial. JAMA 288 (3): 321-33, 2002.
- Chlebowski RT, Hendrix SL, Langer RD, et al.: Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the Women's Health Initiative Randomized Trial. JAMA 289 (24): 3243-53, 2003.
- Beral V; Million Women Study Collaborators: Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet 362 (9382): 419-27, 2003.
- Anderson GL, Limacher M, Assaf AR, et al.: Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA 291 (14): 1701-12, 2004.
- Schuurman AG, van den Brandt PA, Goldbohm RA: Exogenous hormone use and the risk of postmenopausal breast cancer: results from The Netherlands Cohort Study. Cancer Causes Control 6 (5): 416-24, 1995.
- Steinberg KK, Thacker SB, Smith SJ, et al.: A meta-analysis of the effect of estrogen replacement therapy on the risk of breast cancer. JAMA 265 (15): 1985-90, 1991.
- Sellers TA, Mink PJ, Cerhan JR, et al.: The role of hormone replacement therapy in the risk for breast cancer and total mortality in women with a family history of breast cancer. Ann Intern Med 127 (11): 973-80, 1997.
- Stanford JL, Weiss NS, Voigt LF, et al.: Combined estrogen and progestin hormone replacement therapy in relation to risk of breast cancer in middle-aged women. JAMA 274 (2): 137-42, 1995.
- Colditz GA, Egan KM, Stampfer MJ: Hormone replacement therapy and risk of breast cancer: results from epidemiologic studies. Am J Obstet Gynecol 168 (5): 1473-80, 1993.
- Rebbeck TR, Friebel T, Wagner T, et al.: Effect of short-term hormone replacement therapy on breast cancer risk reduction after bilateral prophylactic oophorectomy in BRCA1 and BRCA2 mutation carriers: the PROSE Study Group. J Clin Oncol 23 (31): 7804-10, 2005.
- Kotsopoulos J, Gronwald J, Karlan BY, et al.: Hormone Replacement Therapy After Oophorectomy and Breast Cancer Risk Among BRCA1 Mutation Carriers. JAMA Oncol 4 (8): 1059-1065, 2018.
- Gordhandas S, Norquist BM, Pennington KP, et al.: Hormone replacement therapy after risk reducing salpingo-oophorectomy in patients with BRCA1 or BRCA2 mutations; a systematic review of risks and benefits. Gynecol Oncol 153 (1): 192-200, 2019.
- Helzlsouer KJ, Harris EL, Parshad R, et al.: Familial clustering of breast cancer: possible interaction between DNA repair proficiency and radiation exposure in the development of breast cancer. Int J Cancer 64 (1): 14-7, 1995.
- Helzlsouer KJ, Harris EL, Parshad R, et al.: DNA repair proficiency: potential susceptibility factor for breast cancer. J Natl Cancer Inst 88 (11): 754-5, 1996.
- Abbott DW, Thompson ME, Robinson-Benion C, et al.: BRCA1 expression restores radiation resistance in BRCA1-defective cancer cells through enhancement of transcription-coupled DNA repair. J Biol Chem 274 (26): 18808-12, 1999.
- Abbott DW, Freeman ML, Holt JT: Double-strand break repair deficiency and radiation sensitivity in BRCA2 mutant cancer cells. J Natl Cancer Inst 90 (13): 978-85, 1998.
- Easton DF: Cancer risks in A-T heterozygotes. Int J Radiat Biol 66 (6 Suppl): S177-82, 1994.
- Kleihues P, Schäuble B, zur Hausen A, et al.: Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol 150 (1): 1-13, 1997.
- Pierce LJ, Strawderman M, Narod SA, et al.: Effect of radiotherapy after breast-conserving treatment in women with breast cancer and germline BRCA1/2 mutations. J Clin Oncol 18 (19): 3360-9, 2000.
- Narod SA, Lubinski J, Ghadirian P, et al.: Screening mammography and risk of breast cancer in BRCA1 and BRCA2 mutation carriers: a case-control study. Lancet Oncol 7 (5): 402-6, 2006.
- Andrieu N, Easton DF, Chang-Claude J, et al.: Effect of chest X-rays on the risk of breast cancer among BRCA1/2 mutation carriers in the international BRCA1/2 carrier cohort study: a report from the EMBRACE, GENEPSO, GEO-HEBON, and IBCCS Collaborators' Group. J Clin Oncol 24 (21): 3361-6, 2006.
- Goldfrank D, Chuai S, Bernstein JL, et al.: Effect of mammography on breast cancer risk in women with mutations in BRCA1 or BRCA2. Cancer Epidemiol Biomarkers Prev 15 (11): 2311-3, 2006.
- Gronwald J, Pijpe A, Byrski T, et al.: Early radiation exposures and BRCA1-associated breast cancer in young women from Poland. Breast Cancer Res Treat 112 (3): 581-4, 2008.
- Pijpe A, Andrieu N, Easton DF, et al.: Exposure to diagnostic radiation and risk of breast cancer among carriers of BRCA1/2 mutations: retrospective cohort study (GENE-RAD-RISK). BMJ 345: e5660, 2012.
- Giannakeas V, Lubinski J, Gronwald J, et al.: Mammography screening and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers: a prospective study. Breast Cancer Res Treat 147 (1): 113-8, 2014.
- Drooger J, Akdeniz D, Pignol JP, et al.: Adjuvant radiotherapy for primary breast cancer in BRCA1 and BRCA2 mutation carriers and risk of contralateral breast cancer with special attention to patients irradiated at younger age. Breast Cancer Res Treat 154 (1): 171-80, 2015.
- Reiner AS, Robson ME, Mellemkjær L, et al.: Radiation Treatment, ATM, BRCA1/2, and CHEK2*1100delC Pathogenic Variants and Risk of Contralateral Breast Cancer. J Natl Cancer Inst 112 (12): 1275-1279, 2020.
- Smith-Warner SA, Spiegelman D, Yaun SS, et al.: Alcohol and breast cancer in women: a pooled analysis of cohort studies. JAMA 279 (7): 535-40, 1998.
- Hamajima N, Hirose K, Tajima K, et al.: Alcohol, tobacco and breast cancer--collaborative reanalysis of individual data from 53 epidemiological studies, including 58,515 women with breast cancer and 95,067 women without the disease. Br J Cancer 87 (11): 1234-45, 2002.
- McGuire V, John EM, Felberg A, et al.: No increased risk of breast cancer associated with alcohol consumption among carriers of BRCA1 and BRCA2 mutations ages <50 years. Cancer Epidemiol Biomarkers Prev 15 (8): 1565-7, 2006.
- Dennis J, Ghadirian P, Little J, et al.: Alcohol consumption and the risk of breast cancer among BRCA1 and BRCA2 mutation carriers. Breast 19 (6): 479-83, 2010.
- Cybulski C, Lubinski J, Huzarski T, et al.: Prospective evaluation of alcohol consumption and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res Treat 151 (2): 435-41, 2015.
- Brunet JS, Ghadirian P, Rebbeck TR, et al.: Effect of smoking on breast cancer in carriers of mutant BRCA1 or BRCA2 genes. J Natl Cancer Inst 90 (10): 761-6, 1998.
- Ghadirian P, Lubinski J, Lynch H, et al.: Smoking and the risk of breast cancer among carriers of BRCA mutations. Int J Cancer 110 (3): 413-6, 2004.
- Li H, Terry MB, Antoniou AC, et al.: Alcohol Consumption, Cigarette Smoking, and Risk of Breast Cancer for BRCA1 and BRCA2 Mutation Carriers: Results from The BRCA1 and BRCA2 Cohort Consortium. Cancer Epidemiol Biomarkers Prev 29 (2): 368-378, 2020.
- Zeinomar N, Knight JA, Genkinger JM, et al.: Alcohol consumption, cigarette smoking, and familial breast cancer risk: findings from the Prospective Family Study Cohort (ProF-SC). Breast Cancer Res 21 (1): 128, 2019.
- Lammert J, Lubinski J, Gronwald J, et al.: Physical activity during adolescence and young adulthood and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res Treat 169 (3): 561-571, 2018.
- Kehm RD, Genkinger JM, MacInnis RJ, et al.: Recreational Physical Activity Is Associated with Reduced Breast Cancer Risk in Adult Women at High Risk for Breast Cancer: A Cohort Study of Women Selected for Familial and Genetic Risk. Cancer Res 80 (1): 116-125, 2020.
- Amos CI, Struewing JP: Genetic epidemiology of epithelial ovarian cancer. Cancer 71 (2 Suppl): 566-72, 1993.
- Stratton JF, Pharoah P, Smith SK, et al.: A systematic review and meta-analysis of family history and risk of ovarian cancer. Br J Obstet Gynaecol 105 (5): 493-9, 1998.
- Whittemore AS, Harris R, Itnyre J: Characteristics relating to ovarian cancer risk: collaborative analysis of 12 US case-control studies. II. Invasive epithelial ovarian cancers in white women. Collaborative Ovarian Cancer Group. Am J Epidemiol 136 (10): 1184-203, 1992.
- Brinton LA, Lamb EJ, Moghissi KS, et al.: Ovarian cancer risk after the use of ovulation-stimulating drugs. Obstet Gynecol 103 (6): 1194-203, 2004.
- Gronwald J, Byrski T, Huzarski T, et al.: Influence of selected lifestyle factors on breast and ovarian cancer risk in BRCA1 mutation carriers from Poland. Breast Cancer Res Treat 95 (2): 105-9, 2006.
- McLaughlin JR, Risch HA, Lubinski J, et al.: Reproductive risk factors for ovarian cancer in carriers of BRCA1 or BRCA2 mutations: a case-control study. Lancet Oncol 8 (1): 26-34, 2007.
- Antoniou AC, Rookus M, Andrieu N, et al.: Reproductive and hormonal factors, and ovarian cancer risk for BRCA1 and BRCA2 mutation carriers: results from the International BRCA1/2 Carrier Cohort Study. Cancer Epidemiol Biomarkers Prev 18 (2): 601-10, 2009.
- Narod SA, Sun P, Ghadirian P, et al.: Tubal ligation and risk of ovarian cancer in carriers of BRCA1 or BRCA2 mutations: a case-control study. Lancet 357 (9267): 1467-70, 2001.
- Kotsopoulos J, Lubinski J, Gronwald J, et al.: Factors influencing ovulation and the risk of ovarian cancer in BRCA1 and BRCA2 mutation carriers. Int J Cancer 137 (5): 1136-46, 2015.
- Rodriguez C, Patel AV, Calle EE, et al.: Estrogen replacement therapy and ovarian cancer mortality in a large prospective study of US women. JAMA 285 (11): 1460-5, 2001.
- Riman T, Dickman PW, Nilsson S, et al.: Hormone replacement therapy and the risk of invasive epithelial ovarian cancer in Swedish women. J Natl Cancer Inst 94 (7): 497-504, 2002.
- Lacey JV, Mink PJ, Lubin JH, et al.: Menopausal hormone replacement therapy and risk of ovarian cancer. JAMA 288 (3): 334-41, 2002.
- Anderson GL, Judd HL, Kaunitz AM, et al.: Effects of estrogen plus progestin on gynecologic cancers and associated diagnostic procedures: the Women's Health Initiative randomized trial. JAMA 290 (13): 1739-48, 2003.
- Tortolero-Luna G, Mitchell MF: The epidemiology of ovarian cancer. J Cell Biochem Suppl 23: 200-7, 1995.
- Hankinson SE, Hunter DJ, Colditz GA, et al.: Tubal ligation, hysterectomy, and risk of ovarian cancer. A prospective study. JAMA 270 (23): 2813-8, 1993.
- Rutter JL, Wacholder S, Chetrit A, et al.: Gynecologic surgeries and risk of ovarian cancer in women with BRCA1 and BRCA2 Ashkenazi founder mutations: an Israeli population-based case-control study. J Natl Cancer Inst 95 (14): 1072-8, 2003.
- Kauff ND, Satagopan JM, Robson ME, et al.: Risk-reducing salpingo-oophorectomy in women with a BRCA1 or BRCA2 mutation. N Engl J Med 346 (21): 1609-15, 2002.
- Rebbeck TR, Lynch HT, Neuhausen SL, et al.: Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med 346 (21): 1616-22, 2002.
- Kotsopoulos J, Huzarski T, Gronwald J, et al.: Bilateral Oophorectomy and Breast Cancer Risk in BRCA1 and BRCA2 Mutation Carriers. J Natl Cancer Inst 109 (1): , 2017.
- John EM, Whittemore AS, Harris R, et al.: Characteristics relating to ovarian cancer risk: collaborative analysis of seven U.S. case-control studies. Epithelial ovarian cancer in black women. Collaborative Ovarian Cancer Group. J Natl Cancer Inst 85 (2): 142-7, 1993.
- Narod SA, Risch H, Moslehi R, et al.: Oral contraceptives and the risk of hereditary ovarian cancer. Hereditary Ovarian Cancer Clinical Study Group. N Engl J Med 339 (7): 424-8, 1998.
- Whittemore AS, Balise RR, Pharoah PD, et al.: Oral contraceptive use and ovarian cancer risk among carriers of BRCA1 or BRCA2 mutations. Br J Cancer 91 (11): 1911-5, 2004.
- McGuire V, Felberg A, Mills M, et al.: Relation of contraceptive and reproductive history to ovarian cancer risk in carriers and noncarriers of BRCA1 gene mutations. Am J Epidemiol 160 (7): 613-8, 2004.
- Modan B, Hartge P, Hirsh-Yechezkel G, et al.: Parity, oral contraceptives, and the risk of ovarian cancer among carriers and noncarriers of a BRCA1 or BRCA2 mutation. N Engl J Med 345 (4): 235-40, 2001.
- Soliman PT, Oh JC, Schmeler KM, et al.: Risk factors for young premenopausal women with endometrial cancer. Obstet Gynecol 105 (3): 575-80, 2005.
- Vasen HF, Stormorken A, Menko FH, et al.: MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol 19 (20): 4074-80, 2001.
- Daniels MS: Genetic testing by cancer site: uterus. Cancer J 18 (4): 338-42, 2012 Jul-Aug.
- Dunlop MG, Farrington SM, Nicholl I, et al.: Population carrier frequency of hMSH2 and hMLH1 mutations. Br J Cancer 83 (12): 1643-5, 2000.
- de la Chapelle A: The incidence of Lynch syndrome. Fam Cancer 4 (3): 233-7, 2005.
- Ring KL, Bruegl AS, Allen BA, et al.: Germline multi-gene hereditary cancer panel testing in an unselected endometrial cancer cohort. Mod Pathol 29 (11): 1381-1389, 2016.
- Shu CA, Pike MC, Jotwani AR, et al.: Uterine Cancer After Risk-Reducing Salpingo-oophorectomy Without Hysterectomy in Women With BRCA Mutations. JAMA Oncol 2 (11): 1434-1440, 2016.
- de Jonge MM, de Kroon CD, Jenner DJ, et al.: Endometrial Cancer Risk in Women With Germline BRCA1 or BRCA2 Mutations: Multicenter Cohort Study. J Natl Cancer Inst 113 (9): 1203-1211, 2021.
- McPherson CP, Sellers TA, Potter JD, et al.: Reproductive factors and risk of endometrial cancer. The Iowa Women's Health Study. Am J Epidemiol 143 (12): 1195-202, 1996.
- Dossus L, Allen N, Kaaks R, et al.: Reproductive risk factors and endometrial cancer: the European Prospective Investigation into Cancer and Nutrition. Int J Cancer 127 (2): 442-51, 2010.
- Shapiro S, Kelly JP, Rosenberg L, et al.: Risk of localized and widespread endometrial cancer in relation to recent and discontinued use of conjugated estrogens. N Engl J Med 313 (16): 969-72, 1985.
- Ziel HK, Finkle WD: Increased risk of endometrial carcinoma among users of conjugated estrogens. N Engl J Med 293 (23): 1167-70, 1975.
- Fisher B, Costantino JP, Redmond CK, et al.: Endometrial cancer in tamoxifen-treated breast cancer patients: findings from the National Surgical Adjuvant Breast and Bowel Project (NSABP) B-14. J Natl Cancer Inst 86 (7): 527-37, 1994.
- Pike MC, Peters RK, Cozen W, et al.: Estrogen-progestin replacement therapy and endometrial cancer. J Natl Cancer Inst 89 (15): 1110-6, 1997.
- Fournier A, Dossus L, Mesrine S, et al.: Risks of endometrial cancer associated with different hormone replacement therapies in the E3N cohort, 1992-2008. Am J Epidemiol 180 (5): 508-17, 2014.
- Weiss NS, Sayvetz TA: Incidence of endometrial cancer in relation to the use of oral contraceptives. N Engl J Med 302 (10): 551-4, 1980.
- Soini T, Hurskainen R, Grénman S, et al.: Cancer risk in women using the levonorgestrel-releasing intrauterine system in Finland. Obstet Gynecol 124 (2 Pt 1): 292-9, 2014.
- Lindor NM, McMaster ML, Lindor CJ, et al.: Concise handbook of familial cancer susceptibility syndromes - second edition. J Natl Cancer Inst Monogr (38): 1-93, 2008.
- Vasen HF, Offerhaus GJ, den Hartog Jager FC, et al.: The tumour spectrum in hereditary non-polyposis colorectal cancer: a study of 24 kindreds in the Netherlands. Int J Cancer 46 (1): 31-4, 1990.
- Watson P, Lynch HT: Extracolonic cancer in hereditary nonpolyposis colorectal cancer. Cancer 71 (3): 677-85, 1993.
- Watson P, Vasen HF, Mecklin JP, et al.: The risk of endometrial cancer in hereditary nonpolyposis colorectal cancer. Am J Med 96 (6): 516-20, 1994.
- Aarnio M, Mecklin JP, Aaltonen LA, et al.: Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer 64 (6): 430-3, 1995.
- Raymond VM, Everett JN, Furtado LV, et al.: Adrenocortical carcinoma is a lynch syndrome-associated cancer. J Clin Oncol 31 (24): 3012-8, 2013.
- Raymond VM, Mukherjee B, Wang F, et al.: Elevated risk of prostate cancer among men with Lynch syndrome. J Clin Oncol 31 (14): 1713-8, 2013.
- Suspiro A, Fidalgo P, Cravo M, et al.: The Muir-Torre syndrome: a rare variant of hereditary nonpolyposis colorectal cancer associated with hMSH2 mutation. Am J Gastroenterol 93 (9): 1572-4, 1998.
- Kerber RA, Slattery ML: Comparison of self-reported and database-linked family history of cancer data in a case-control study. Am J Epidemiol 146 (3): 244-8, 1997.
- Parent ME, Ghadirian P, Lacroix A, et al.: The reliability of recollections of family history: implications for the medical provider. J Cancer Educ 12 (2): 114-20, 1997 Summer.
- Ready K, Litton JK, Arun BK: Clinical application of breast cancer risk assessment models. Future Oncol 6 (3): 355-65, 2010.
- Amir E, Freedman OC, Seruga B, et al.: Assessing women at high risk of breast cancer: a review of risk assessment models. J Natl Cancer Inst 102 (10): 680-91, 2010.
- Rosner BA, Colditz GA, Webb PM, et al.: Mathematical models of ovarian cancer incidence. Epidemiology 16 (4): 508-15, 2005.
- Pfeiffer RM, Park Y, Kreimer AR, et al.: Risk prediction for breast, endometrial, and ovarian cancer in white women aged 50 y or older: derivation and validation from population-based cohort studies. PLoS Med 10 (7): e1001492, 2013.
- Gail MH, Mai PL: Comparing breast cancer risk assessment models. J Natl Cancer Inst 102 (10): 665-8, 2010.
- Quante AS, Whittemore AS, Shriver T, et al.: Breast cancer risk assessment across the risk continuum: genetic and nongenetic risk factors contributing to differential model performance. Breast Cancer Res 14 (6): R144, 2012.
- Gail MH, Brinton LA, Byar DP, et al.: Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst 81 (24): 1879-86, 1989.
- Colditz GA, Rosner B: Cumulative risk of breast cancer to age 70 years according to risk factor status: data from the Nurses' Health Study. Am J Epidemiol 152 (10): 950-64, 2000.
- Claus EB, Risch N, Thompson WD: Autosomal dominant inheritance of early-onset breast cancer. Implications for risk prediction. Cancer 73 (3): 643-51, 1994.
- Tyrer J, Duffy SW, Cuzick J: A breast cancer prediction model incorporating familial and personal risk factors. Stat Med 23 (7): 1111-30, 2004.
- Parmigiani G, Berry D, Aguilar O: Determining carrier probabilities for breast cancer-susceptibility genes BRCA1 and BRCA2. Am J Hum Genet 62 (1): 145-58, 1998.
- Antoniou AC, Pharoah PD, McMullan G, et al.: A comprehensive model for familial breast cancer incorporating BRCA1, BRCA2 and other genes. Br J Cancer 86 (1): 76-83, 2002.
- Antoniou AC, Pharoah PP, Smith P, et al.: The BOADICEA model of genetic susceptibility to breast and ovarian cancer. Br J Cancer 91 (8): 1580-90, 2004.
- Antoniou AC, Cunningham AP, Peto J, et al.: The BOADICEA model of genetic susceptibility to breast and ovarian cancers: updates and extensions. Br J Cancer 98 (8): 1457-66, 2008.
- Anothaisintawee T, Teerawattananon Y, Wiratkapun C, et al.: Risk prediction models of breast cancer: a systematic review of model performances. Breast Cancer Res Treat 133 (1): 1-10, 2012.
- Amir E, Evans DG, Shenton A, et al.: Evaluation of breast cancer risk assessment packages in the family history evaluation and screening programme. J Med Genet 40 (11): 807-14, 2003.
- Laitman Y, Simeonov M, Keinan-Boker L, et al.: Breast cancer risk prediction accuracy in Jewish Israeli high-risk women using the BOADICEA and IBIS risk models. Genet Res (Camb) 95 (6): 174-7, 2013.
- MacInnis RJ, Bickerstaffe A, Apicella C, et al.: Prospective validation of the breast cancer risk prediction model BOADICEA and a batch-mode version BOADICEACentre. Br J Cancer 109 (5): 1296-301, 2013.
- Terry MB, Liao Y, Whittemore AS, et al.: 10-year performance of four models of breast cancer risk: a validation study. Lancet Oncol 20 (4): 504-517, 2019.
- Claus EB, Risch N, Thompson WD: The calculation of breast cancer risk for women with a first degree family history of ovarian cancer. Breast Cancer Res Treat 28 (2): 115-20, 1993.
- Bondy ML, Lustbader ED, Halabi S, et al.: Validation of a breast cancer risk assessment model in women with a positive family history. J Natl Cancer Inst 86 (8): 620-5, 1994.
- Spiegelman D, Colditz GA, Hunter D, et al.: Validation of the Gail et al. model for predicting individual breast cancer risk. J Natl Cancer Inst 86 (8): 600-7, 1994.
- Rockhill B, Spiegelman D, Byrne C, et al.: Validation of the Gail et al. model of breast cancer risk prediction and implications for chemoprevention. J Natl Cancer Inst 93 (5): 358-66, 2001.
- Costantino JP, Gail MH, Pee D, et al.: Validation studies for models projecting the risk of invasive and total breast cancer incidence. J Natl Cancer Inst 91 (18): 1541-8, 1999.
- Bondy ML, Newman LA: Breast cancer risk assessment models: applicability to African-American women. Cancer 97 (1 Suppl): 230-5, 2003.
- Schonfeld SJ, Pee D, Greenlee RT, et al.: Effect of changing breast cancer incidence rates on the calibration of the Gail model. J Clin Oncol 28 (14): 2411-7, 2010.
- Gail MH, Costantino JP, Pee D, et al.: Projecting individualized absolute invasive breast cancer risk in African American women. J Natl Cancer Inst 99 (23): 1782-92, 2007.
- Gail MH: Discriminatory accuracy from single-nucleotide polymorphisms in models to predict breast cancer risk. J Natl Cancer Inst 100 (14): 1037-41, 2008.
- Gail MH: Value of adding single-nucleotide polymorphism genotypes to a breast cancer risk model. J Natl Cancer Inst 101 (13): 959-63, 2009.
- Barlow WE, White E, Ballard-Barbash R, et al.: Prospective breast cancer risk prediction model for women undergoing screening mammography. J Natl Cancer Inst 98 (17): 1204-14, 2006.
- Tice JA, Cummings SR, Ziv E, et al.: Mammographic breast density and the Gail model for breast cancer risk prediction in a screening population. Breast Cancer Res Treat 94 (2): 115-22, 2005.
- Lee AJ, Cunningham AP, Tischkowitz M, et al.: Incorporating truncating variants in PALB2, CHEK2, and ATM into the BOADICEA breast cancer risk model. Genet Med 18 (12): 1190-1198, 2016.
- MacInnis RJ, Liao Y, Knight JA, et al.: Considerations When Using Breast Cancer Risk Models for Women with Negative BRCA1/BRCA2 Mutation Results. J Natl Cancer Inst 112 (4): 418-422, 2020.
- Barnetson RA, Tenesa A, Farrington SM, et al.: Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med 354 (26): 2751-63, 2006.
- Kastrinos F, Uno H, Ukaegbu C, et al.: Development and Validation of the PREMM5 Model for Comprehensive Risk Assessment of Lynch Syndrome. J Clin Oncol 35 (19): 2165-2172, 2017.
- Chen S, Wang W, Lee S, et al.: Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA 296 (12): 1479-87, 2006.
- Khan O, Blanco A, Conrad P, et al.: Performance of Lynch syndrome predictive models in a multi-center US referral population. Am J Gastroenterol 106 (10): 1822-7; quiz 1828, 2011.
- Mercado RC, Hampel H, Kastrinos F, et al.: Performance of PREMM(1,2,6), MMRpredict, and MMRpro in detecting Lynch syndrome among endometrial cancer cases. Genet Med 14 (7): 670-80, 2012.
- Couch FJ, DeShano ML, Blackwood MA, et al.: BRCA1 mutations in women attending clinics that evaluate the risk of breast cancer. N Engl J Med 336 (20): 1409-15, 1997.
- Shattuck-Eidens D, Oliphant A, McClure M, et al.: BRCA1 sequence analysis in women at high risk for susceptibility mutations. Risk factor analysis and implications for genetic testing. JAMA 278 (15): 1242-50, 1997.
- Frank TS, Manley SA, Olopade OI, et al.: Sequence analysis of BRCA1 and BRCA2: correlation of mutations with family history and ovarian cancer risk. J Clin Oncol 16 (7): 2417-25, 1998.
- Frank TS, Deffenbaugh AM, Reid JE, et al.: Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J Clin Oncol 20 (6): 1480-90, 2002.
- Evans DG, Eccles DM, Rahman N, et al.: A new scoring system for the chances of identifying a BRCA1/2 mutation outperforms existing models including BRCAPRO. J Med Genet 41 (6): 474-80, 2004.
- Apicella C, Dowty JG, Dite GS, et al.: Validation study of the LAMBDA model for predicting the BRCA1 or BRCA2 mutation carrier status of North American Ashkenazi Jewish women. Clin Genet 72 (2): 87-97, 2007.
- Risch HA, McLaughlin JR, Cole DE, et al.: Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet 68 (3): 700-10, 2001.
- Malone KE, Daling JR, Doody DR, et al.: Prevalence and predictors of BRCA1 and BRCA2 mutations in a population-based study of breast cancer in white and black American women ages 35 to 64 years. Cancer Res 66 (16): 8297-308, 2006.
- Struewing JP, Abeliovich D, Peretz T, et al.: The carrier frequency of the BRCA1 185delAG mutation is approximately 1 percent in Ashkenazi Jewish individuals. Nat Genet 11 (2): 198-200, 1995.
- Oddoux C, Struewing JP, Clayton CM, et al.: The carrier frequency of the BRCA2 6174delT mutation among Ashkenazi Jewish individuals is approximately 1%. Nat Genet 14 (2): 188-90, 1996.
- Warner E, Foulkes W, Goodwin P, et al.: Prevalence and penetrance of BRCA1 and BRCA2 gene mutations in unselected Ashkenazi Jewish women with breast cancer. J Natl Cancer Inst 91 (14): 1241-7, 1999.
- Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Anglian Breast Cancer Study Group. Br J Cancer 83 (10): 1301-8, 2000.
- Euhus DM, Smith KC, Robinson L, et al.: Pretest prediction of BRCA1 or BRCA2 mutation by risk counselors and the computer model BRCAPRO. J Natl Cancer Inst 94 (11): 844-51, 2002.
- de la Hoya M, Díez O, Pérez-Segura P, et al.: Pre-test prediction models of BRCA1 or BRCA2 mutation in breast/ovarian families attending familial cancer clinics. J Med Genet 40 (7): 503-10, 2003.
- Katki HA: Incorporating medical interventions into carrier probability estimation for genetic counseling. BMC Med Genet 8: 13, 2007.
- Weitzel JN, Lagos VI, Cullinane CA, et al.: Limited family structure and BRCA gene mutation status in single cases of breast cancer. JAMA 297 (23): 2587-95, 2007.
- Jervis S, Song H, Lee A, et al.: A risk prediction algorithm for ovarian cancer incorporating BRCA1, BRCA2, common alleles and other familial effects. J Med Genet 52 (7): 465-75, 2015.
- Oros KK, Ghadirian P, Maugard CM, et al.: Application of BRCA1 and BRCA2 mutation carrier prediction models in breast and/or ovarian cancer families of French Canadian descent. Clin Genet 70 (4): 320-9, 2006.
- Vogel KJ, Atchley DP, Erlichman J, et al.: BRCA1 and BRCA2 genetic testing in Hispanic patients: mutation prevalence and evaluation of the BRCAPRO risk assessment model. J Clin Oncol 25 (29): 4635-41, 2007.
- Kurian AW, Gong GD, John EM, et al.: Performance of prediction models for BRCA mutation carriage in three racial/ethnic groups: findings from the Northern California Breast Cancer Family Registry. Cancer Epidemiol Biomarkers Prev 18 (4): 1084-91, 2009.
- Kurian AW, Gong GD, Chun NM, et al.: Performance of BRCA1/2 mutation prediction models in Asian Americans. J Clin Oncol 26 (29): 4752-8, 2008.
- Berry DA, Parmigiani G, Sanchez J, et al.: Probability of carrying a mutation of breast-ovarian cancer gene BRCA1 based on family history. J Natl Cancer Inst 89 (3): 227-38, 1997.
- Barcenas CH, Hosain GM, Arun B, et al.: Assessing BRCA carrier probabilities in extended families. J Clin Oncol 24 (3): 354-60, 2006.
- Kang HH, Williams R, Leary J, et al.: Evaluation of models to predict BRCA germline mutations. Br J Cancer 95 (7): 914-20, 2006.
- Antoniou AC, Hardy R, Walker L, et al.: Predicting the likelihood of carrying a BRCA1 or BRCA2 mutation: validation of BOADICEA, BRCAPRO, IBIS, Myriad and the Manchester scoring system using data from UK genetics clinics. J Med Genet 45 (7): 425-31, 2008.
- Fischer C, Kuchenbäcker K, Engel C, et al.: Evaluating the performance of the breast cancer genetic risk models BOADICEA, IBIS, BRCAPRO and Claus for predicting BRCA1/2 mutation carrier probabilities: a study based on 7352 families from the German Hereditary Breast and Ovarian Cancer Consortium. J Med Genet 50 (6): 360-7, 2013.
- Biswas S, Tankhiwale N, Blackford A, et al.: Assessing the added value of breast tumor markers in genetic risk prediction model BRCAPRO. Breast Cancer Res Treat 133 (1): 347-55, 2012.
- Tai YC, Chen S, Parmigiani G, et al.: Incorporating tumor immunohistochemical markers in BRCA1 and BRCA2 carrier prediction. Breast Cancer Res 10 (2): 401, 2008.
- Ready KJ, Vogel KJ, Atchley DP, et al.: Accuracy of the BRCAPRO model among women with bilateral breast cancer. Cancer 115 (4): 725-30, 2009.
- Huo D, Senie RT, Daly M, et al.: Prediction of BRCA Mutations Using the BRCAPRO Model in Clinic-Based African American, Hispanic, and Other Minority Families in the United States. J Clin Oncol 27 (8): 1184-90, 2009.
- Daniels MS, Babb SA, King RH, et al.: Underestimation of risk of a BRCA1 or BRCA2 mutation in women with high-grade serous ovarian cancer by BRCAPRO: a multi-institution study. J Clin Oncol 32 (12): 1249-55, 2014.
- Robson ME, Storm CD, Weitzel J, et al.: American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol 28 (5): 893-901, 2010.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 2.2024. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed October 31, 2023.
- Statement of the American Society of Human Genetics on genetic testing for breast and ovarian cancer predisposition. Am J Hum Genet 55 (5): i-iv, 1994.
- Hampel H, Bennett RL, Buchanan A, et al.: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 17 (1): 70-87, 2015.
- Owens DK, Davidson KW, Krist AH, et al.: Risk Assessment, Genetic Counseling, and Genetic Testing for BRCA-Related Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 322 (7): 652-665, 2019.
- Lancaster JM, Powell CB, Kauff ND, et al.: Society of Gynecologic Oncologists Education Committee statement on risk assessment for inherited gynecologic cancer predispositions. Gynecol Oncol 107 (2): 159-62, 2007.
- Manahan ER, Kuerer HM, Sebastian M, et al.: Consensus Guidelines on Genetic` Testing for Hereditary Breast Cancer from the American Society of Breast Surgeons. Ann Surg Oncol 26 (10): 3025-3031, 2019.
- Beitsch PD, Whitworth PW, Hughes K, et al.: Underdiagnosis of Hereditary Breast Cancer: Are Genetic Testing Guidelines a Tool or an Obstacle? J Clin Oncol 37 (6): 453-460, 2019.
- Cropper C, Woodson A, Arun B, et al.: Evaluating the NCCN Clinical Criteria for Recommending BRCA1 and BRCA2 Genetic Testing in Patients With Breast Cancer. J Natl Compr Canc Netw 15 (6): 797-803, 2017.
- Grindedal EM, Heramb C, Karsrud I, et al.: Current guidelines for BRCA testing of breast cancer patients are insufficient to detect all mutation carriers. BMC Cancer 17 (1): 438, 2017.
- Yadav S, Hu C, Hart SN, et al.: Evaluation of Germline Genetic Testing Criteria in a Hospital-Based Series of Women With Breast Cancer. J Clin Oncol 38 (13): 1409-1418, 2020.
- Kurian AW, Bernhisel R, Larson K, et al.: Prevalence of Pathogenic Variants in Cancer Susceptibility Genes Among Women With Postmenopausal Breast Cancer. JAMA 323 (10): 995-997, 2020.
- Couch FJ, Nathanson KL, Offit K: Two decades after BRCA: setting paradigms in personalized cancer care and prevention. Science 343 (6178): 1466-70, 2014.
- Theobald KA, Susswein LR, Marshall ML, et al.: Utility of Expedited Hereditary Cancer Testing in the Surgical Management of Patients with a New Breast Cancer Diagnosis. Ann Surg Oncol 25 (12): 3556-3562, 2018.
- Gornick MC, Kurian AW, An LC, et al.: Knowledge regarding and patterns of genetic testing in patients newly diagnosed with breast cancer participating in the iCanDecide trial. Cancer 124 (20): 4000-4009, 2018.
- Chiba A, Hoskin TL, Hallberg EJ, et al.: Impact that Timing of Genetic Mutation Diagnosis has on Surgical Decision Making and Outcome for BRCA1/BRCA2 Mutation Carriers with Breast Cancer. Ann Surg Oncol 23 (10): 3232-8, 2016.
- Obermair A, Youlden DR, Baade PD, et al.: The impact of risk-reducing hysterectomy and bilateral salpingo-oophorectomy on survival in patients with a history of breast cancer--a population-based data linkage study. Int J Cancer 134 (9): 2211-22, 2014.
- Byrski T, Dent R, Blecharz P, et al.: Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. Breast Cancer Res 14 (4): R110, 2012.
- Robson M, Im SA, Senkus E, et al.: Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med 377 (6): 523-533, 2017.
- Heymann S, Delaloge S, Rahal A, et al.: Radio-induced malignancies after breast cancer postoperative radiotherapy in patients with Li-Fraumeni syndrome. Radiat Oncol 5: 104, 2010.
- Bougeard G, Renaux-Petel M, Flaman JM, et al.: Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J Clin Oncol 33 (21): 2345-52, 2015.
- Rebbeck TR, Kauff ND, Domchek SM: Meta-analysis of risk reduction estimates associated with risk-reducing salpingo-oophorectomy in BRCA1 or BRCA2 mutation carriers. J Natl Cancer Inst 101 (2): 80-7, 2009.
- Kotsopoulos J, Lubinski J, Lynch HT, et al.: Oophorectomy after menopause and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. Cancer Epidemiol Biomarkers Prev 21 (7): 1089-96, 2012.
- Cass I, Baldwin RL, Varkey T, et al.: Improved survival in women with BRCA-associated ovarian carcinoma. Cancer 97 (9): 2187-95, 2003.
- Tan DS, Rothermundt C, Thomas K, et al.: "BRCAness" syndrome in ovarian cancer: a case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J Clin Oncol 26 (34): 5530-6, 2008.
- Moore K, Colombo N, Scambia G, et al.: Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med 379 (26): 2495-2505, 2018.
- Practice Bulletin No. 149: Endometrial cancer. Obstet Gynecol 125 (4): 1006-26, 2015.
- Ott PA, Bang YJ, Berton-Rigaud D, et al.: Safety and Antitumor Activity of Pembrolizumab in Advanced Programmed Death Ligand 1-Positive Endometrial Cancer: Results From the KEYNOTE-028 Study. J Clin Oncol 35 (22): 2535-2541, 2017.
- Walsh T, Casadei S, Lee MK, et al.: Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A 108 (44): 18032-7, 2011.
- Frey MK, Kim SH, Bassett RY, et al.: Rescreening for genetic mutations using multi-gene panel testing in patients who previously underwent non-informative genetic screening. Gynecol Oncol 139 (2): 211-5, 2015.
- Desmond A, Kurian AW, Gabree M, et al.: Clinical Actionability of Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Risk Assessment. JAMA Oncol 1 (7): 943-51, 2015.
- Couch FJ, Shimelis H, Hu C, et al.: Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol 3 (9): 1190-1196, 2017.
- Weitzel JN, Neuhausen SL, Adamson A, et al.: Pathogenic and likely pathogenic variants in PALB2, CHEK2, and other known breast cancer susceptibility genes among 1054 BRCA-negative Hispanics with breast cancer. Cancer 125 (16): 2829-2836, 2019.
- Frey MK, Kopparam RV, Ni Zhou Z, et al.: Prevalence of nonfounder BRCA1/2 mutations in Ashkenazi Jewish patients presenting for genetic testing at a hereditary breast and ovarian cancer center. Cancer 125 (5): 690-697, 2019.
- Eoh KJ, Kim JE, Park HS, et al.: Detection of Germline Mutations in Patients with Epithelial Ovarian Cancer Using Multi-gene Panels: Beyond BRCA1/2. Cancer Res Treat 50 (3): 917-925, 2018.
- Tung N, Lin NU, Kidd J, et al.: Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J Clin Oncol 34 (13): 1460-8, 2016.
- Buys SS, Sandbach JF, Gammon A, et al.: A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 123 (10): 1721-1730, 2017.
- Pritzlaff M, Summerour P, McFarland R, et al.: Male breast cancer in a multi-gene panel testing cohort: insights and unexpected results. Breast Cancer Res Treat 161 (3): 575-586, 2017.
- Yadav S, Reeves A, Campian S, et al.: Outcomes of retesting BRCA negative patients using multigene panels. Fam Cancer 16 (3): 319-328, 2017.
- Crawford B, Adams SB, Sittler T, et al.: Multi-gene panel testing for hereditary cancer predisposition in unsolved high-risk breast and ovarian cancer patients. Breast Cancer Res Treat 163 (2): 383-390, 2017.
- Kurian AW, Ward KC, Howlader N, et al.: Genetic Testing and Results in a Population-Based Cohort of Breast Cancer Patients and Ovarian Cancer Patients. J Clin Oncol 37 (15): 1305-1315, 2019.
- Carter NJ, Marshall ML, Susswein LR, et al.: Germline pathogenic variants identified in women with ovarian tumors. Gynecol Oncol 151 (3): 481-488, 2018.
- Dorling L, Carvalho S, Allen J, et al.: Breast Cancer Risk Genes - Association Analysis in More than 113,000 Women. N Engl J Med 384 (5): 428-439, 2021.
- Hu C, Hart SN, Gnanaolivu R, et al.: A Population-Based Study of Genes Previously Implicated in Breast Cancer. N Engl J Med 384 (5): 440-451, 2021.
- Shimelis H, LaDuca H, Hu C, et al.: Triple-Negative Breast Cancer Risk Genes Identified by Multigene Hereditary Cancer Panel Testing. J Natl Cancer Inst 110 (8): 855-862, 2018.
- Palmer JR, Polley EC, Hu C, et al.: Contribution of Germline Predisposition Gene Mutations to Breast Cancer Risk in African American Women. J Natl Cancer Inst 112 (12): 1213-1221, 2020.
- Díaz-Zabala H, Guo X, Ping J, et al.: Evaluating breast cancer predisposition genes in women of African ancestry. Genet Med 24 (7): 1468-1475, 2022.
- Southey MC, Goldgar DE, Winqvist R, et al.: PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J Med Genet 53 (12): 800-811, 2016.
- Kurian AW, Ward KC, Hamilton AS, et al.: Uptake, Results, and Outcomes of Germline Multiple-Gene Sequencing After Diagnosis of Breast Cancer. JAMA Oncol 4 (8): 1066-1072, 2018.
Penetrance of Inherited Susceptibility to Hereditary Breast and/or Gynecologic Cancers
The proportion of individuals carrying a pathogenic variant who will manifest a certain disease is referred to as penetrance. In general, common genetic variants that are associated with cancer susceptibility have a lower penetrance than rare genetic variants. This is depicted in Figure 4. For adult-onset diseases, penetrance is usually described by the individual carrier's age, sex, and organ site. For example, the penetrance for breast cancer in female carriers of BRCA1 pathogenic variants is often quoted by age 50 years and by age 70 years. Of the numerous methods for estimating penetrance, none are without potential biases, and determining an individual carrier's risk of cancer involves some level of imprecision.
 Figure 4. Genetic architecture of cancer risk. This graph depicts the general finding of a low relative risk associated with common, low-penetrance genetic variants, such as single-nucleotide polymorphisms identified in genome-wide association studies, and a higher relative risk associated with rare, high-penetrance genetic variants, such as pathogenic variants in the BRCA1/BRCA2 genes associated with hereditary breast and ovarian cancer and the mismatch repair genes associated with Lynch syndrome.
Figure 4. Genetic architecture of cancer risk. This graph depicts the general finding of a low relative risk associated with common, low-penetrance genetic variants, such as single-nucleotide polymorphisms identified in genome-wide association studies, and a higher relative risk associated with rare, high-penetrance genetic variants, such as pathogenic variants in the BRCA1/BRCA2 genes associated with hereditary breast and ovarian cancer and the mismatch repair genes associated with Lynch syndrome.
Throughout this summary, we discuss studies that report on relative and absolute risks (ARs). These are two important but different concepts. Relative risk (RR) refers to an estimate of risk relative to another group (e.g., risk of an outcome like breast cancer for women who are exposed to a risk factor relative to the risk of breast cancer for women who are unexposed to the same risk factor). RR measures that are greater than 1 mean that the risk for those captured in the numerator (i.e., the exposed) is higher than the risk for those captured in the denominator (i.e., the unexposed). RR measures that are less than 1 mean that the risk for those captured in the numerator (i.e., the exposed) is lower than the risk for those captured in the denominator (i.e., the unexposed). Measures with similar relative interpretations include the odds ratio (OR), hazard ratio, and risk ratio.
AR measures consider the number of people who have a particular outcome, the number of people in a population who could have the outcome, and person-time (the period of time during which an individual was at risk of having the outcome). AR measures also reflect the absolute burden of an outcome in a population. Absolute measures include risks and rates and can be expressed over a specific time frame (e.g., 1 year, 5 years) or overall lifetime. Cumulative risk is a measure of risk that occurs over a defined time period. For example, overall lifetime risk is a type of cumulative risk that is usually calculated on the basis of a given life expectancy (e.g., 80 or 90 years). Cumulative risk can also be presented over other time frames (e.g., up to age 50 years).
Large RR measures do not mean that there will be large effects in the actual number of individuals at a population level because the disease outcome may be quite rare. For example, the RR for smoking is much higher for lung cancer than for heart disease, but the absolute difference between smokers and nonsmokers is greater for heart disease, the more-common outcome, than for lung cancer, the more-rare outcome.
Therefore, in evaluating the effect of exposures and biological markers on disease prevention across the continuum, it is important to recognize the differences between relative and absolute effects in weighing the overall impact of a given risk factor. For example, the magnitude is in the range of 30% (e.g., ORs or RRs of 1.3) for many breast cancer risk factors, which means that women with a risk factor (e.g., alcohol consumption, late age at first birth, oral contraceptive use, postmenopausal body size) have a 30% relative increase in breast cancer in comparison with what they would have if they did not have that risk factor. But the absolute increase in risk is based on the underlying AR of disease. Figure 5 and Table 3 show the impact of a RR factor in the range of 1.3 on AR. As shown, women with a family history of breast cancer have a much higher benefit from risk factor reduction on an absolute scale.
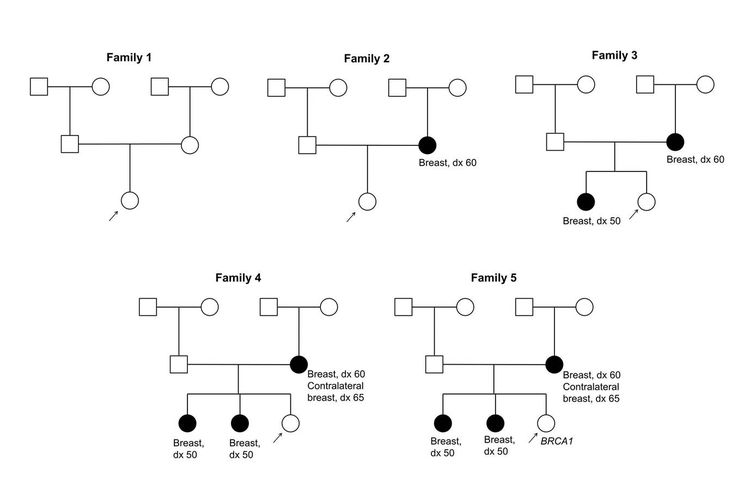 Figure 5. These five pedigrees depict probands with varying degrees of family history. Table 3 accompanies this figure.
Figure 5. These five pedigrees depict probands with varying degrees of family history. Table 3 accompanies this figure.
| Family History | Lifetime Risk (%) | Lifetime Risk After Risk Factor Modification (%) | Absolute Risk Difference (%) | Relative Risk |
|---|---|---|---|---|
| aRefer to Figure 5, which accompanies this table. | ||||
| Low (Family 1) | 10.9 | 8.4 | 2.50 | 1.29 (29% increased risk) |
| Moderate (Family 2) | 21.6 | 16.8 | 4.80 | 1.28 (28% increased risk) |
| Moderate/high (Family 3) | 27.1 | 21.3 | 5.80 | 1.27 (27% increased risk) |
| High (Family 4) | 32.0 | 25.3 | 6.70 | 1.26 (26% increased risk) |
| BRCA1 pathogenic variant (Family 5) | 53.7 | 44.2 | 9.50 | 1.21 (21% increased risk) |
With the increasing use of multigene panel tests, a framework for cancer risk management among individuals with pathogenic variants detected in novel genes has been described that incorporates data on age-specific, lifetime, and absolute cancer risks. The framework suggests initiating screening in these individuals at the age when their 5-year cancer risk approaches that at which screening is routinely initiated for women in the general population (approximately 1% for breast cancer in the United States). As a result, the age at which to begin screening will vary depending on the gene. (Refer to the Multigene [panel] testing section of this summary for more information on multigene panel tests.)
References
- Quante AS, Herz J, Whittemore AS, et al.: Assessing absolute changes in breast cancer risk due to modifiable risk factors. Breast Cancer Res Treat 152 (1): 193-7, 2015.
- Tung N, Domchek SM, Stadler Z, et al.: Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat Rev Clin Oncol 13 (9): 581-8, 2016.
Genes Associated With Breast and/or Gynecologic Cancer Susceptibility
Several genes are found to be associated with the development of breast and/or gynecologic cancers. These genes are categorized as high-penetrance, moderate-penetrance, and low-penetrance in this summary. The high- and moderate-penetrance genes are summarized in Table 4. Low-penetrance genes and loci primarily include polymorphisms that have been associated with cancer susceptibility. (Refer to the High-Penetrance Breast and/or Gynecologic Cancer Susceptibility Genes, Moderate-Penetrance Genes Associated With Breast and/or Gynecologic Cancer, and Single Nucleotide Variant–Associated Cancer Risks sections of this summary for more information.)
| Cancer Susceptibilitya | Moderate-Penetrance Genesb | High-Penetrance Genes |
|---|---|---|
| aOther cancers may be associated with the genes in this table. | ||
| bOther genes discussed in the Moderate-Penetrance Genes Associated With Breast and/or Gynecologic Cancer section of this summary but for which penetrance is unknown include CASP8, TGFB1, Abraxas, RECQL, and SMARCA4. | ||
| Breast cancer | ATM, BRIP1, CHEK2, FANCD2, RAD51C | BRCA1, BRCA2, CDH1, PALB2, PTEN, STK11, TP53 |
| Ovarian cancer | ATM, BRIP1, EPCAM, MLH1, MSH2, MSH6, RAD51C | BRCA1, BRCA2 |
| Endometrial cancer | EPCAM, MLH1, MSH2, MSH6, PMS2, PTEN | |
High-Penetrance Breast and/or Gynecologic Cancer Susceptibility Genes
BRCA1 and BRCA2
For more information about BRCA1 and BRCA2 pathogenic variants and BRCA-associated cancer risks, see BRCA1 and BRCA2: Cancer Risks and Management.
Lynch Syndrome
Lynch syndrome is characterized by autosomal dominant inheritance of susceptibility to predominantly right-sided colon cancer, endometrial cancer, ovarian cancer, and other extracolonic cancers (including cancer of the renal pelvis, ureter, small bowel, and pancreas), multiple primary cancers, and a young age of onset of cancer. The condition is caused by germline variants in the mismatch repair (MMR) genes, which are involved in repair of DNA mismatch variants. The MLH1 and MSH2 genes are the most common susceptibility genes for Lynch syndrome, accounting for 80% to 90% of observed pathogenic variants, followed by MSH6 and PMS2. (Refer to the Lynch Syndrome section in Genetics of Colorectal Cancer for more information about this syndrome.)
After colorectal cancer, endometrial cancer is the second hallmark cancer of a family with Lynch syndrome. Even in the original Family G, described by Dr. Aldred Scott Warthin, numerous family members were noted to have extracolonic cancers including endometrial cancer. Although the first version of the Amsterdam criteria did not include endometrial cancer, in 1999, the Amsterdam criteria were revised to include endometrial cancer as extracolonic tumors associated with Lynch syndrome to identify families at risk. In addition, the Bethesda guidelines in 1997 (revised in 2004) did include endometrial and ovarian cancers as Lynch syndrome–related cancers to prompt tumor testing for Lynch syndrome.
The lifetime risk of ovarian carcinoma in females with Lynch syndrome is estimated to be as high as 12%, and the reported relative risk (RR) of ovarian cancer has ranged from 3.6 to 13, based on families ascertained from high-risk clinics with known or suspected Lynch syndrome. There may be differences in ovarian cancer risk depending on the Lynch syndrome–associated pathogenic variant. In PMS2-associated Lynch syndrome, one study of 284 families was unable to identify an increased risk of ovarian cancer. Another prospective registry of 3,119 Lynch syndrome–pathogenic variant carriers described the cumulative risk of ovarian cancer to range from 10% to 17% in MLH1, MSH2, and MSH6 carriers. In contrast, 0 of 67 women with a pathogenic variant in PMS2 developed ovarian cancer in 303 follow-up years. Overall, there are too few cases of PMS2 pathogenic variant carriers to make definitive recommendations for ovarian cancer management. Characteristics of Lynch syndrome–associated ovarian cancers may include overrepresentation of the International Federation of Gynecology and Obstetrics stages I and II at diagnosis (reported as 81.5%), underrepresentation of serous subtypes (reported as 22.9%), and a better 10-year survival (reported as 80.6%) than reported both in population-based series and in carriers of BRCA pathogenic variants.
The issue of breast cancer risk in Lynch syndrome has been controversial.
Retrospective studies have been inconsistent, but several have demonstrated microsatellite instability in a proportion of breast cancers from individuals with Lynch syndrome; one of these studies evaluated breast cancer risk in individuals with Lynch syndrome and found that it is not elevated. However, the largest prospective study to date of 446 unaffected carriers of pathogenic variants from the Colon Cancer Family Registry who were followed for up to 10 years reported an elevated SIR of 3.95 for breast cancer (95% CI, 1.59–8.13; P = .001). The same group subsequently analyzed data on 764 carriers of MMR gene pathogenic variants with a prior diagnosis of colorectal cancer. Results showed that the 10-year risk of breast cancer following colorectal cancer was 2% (95% CI, 1%–4%) and that the SIR was 1.76 (95% CI, 1.07–2.59). A series from the United Kingdom composed of clinically referred Lynch syndrome kindreds, with efforts to correct for ascertainment, showed a twofold increased risk of breast cancer in 157 MLH1 carriers but not in carriers of other MMR variants. Results from a meta-analysis of breast cancer risk in Lynch syndrome among 15 studies with molecular tumor testing results revealed that 62 of 122 breast cancers (51%; 95% CI, 42%–60%) in MMR pathogenic variant carriers were MMR-deficient. In addition, breast cancer risk estimates among a total of 21 studies showed an increased risk of twofold to 18-fold in eight studies that compared MMR variant carriers with noncarriers, while 13 studies did not observe statistical evidence for an association of breast cancer risk with Lynch syndrome.
A number of subsequent studies have suggested the presence of higher breast cancer risks than previously published, although this has not been consistently observed. Through a study of 325 Canadian families with Lynch syndrome, primarily encompassing MLH1 and MSH2 carriers, the lifetime cumulative risk for breast cancer among MSH2 carriers was reported to be 22%. Similarly, breast cancer risks were elevated in a study of 423 women with Lynch syndrome, with substantially higher risks among those with MSH6 and PMS2 pathogenic variants, compared with MLH1 and MSH2 pathogenic variants. In fact, breast cancer risk to age 60 years was 37.7% for PMS2, 31.1% for MSH6, 16.1% for MSH2, and 15.5% for MLH1. These findings are consistent with another study of 528 patients with Lynch syndrome–associated pathogenic variants (including MLH1, MSH2, MSH6, PMS2, and EPCAM) in which PMS2 and MSH6 variants were much more frequent among patients with only breast cancer, compared with those with only colorectal cancer (P = 2.3 x 10-5). Additional data to support an association of MSH6 with breast cancer were provided through a study of over 10,000 cancer patients across the United States who had genetic testing. Findings indicated that MSH6 was associated with breast cancer with an odds ratio (OR) of 2.59 (95% CI, 1.35–5.44). Taken together, these studies highlight how the risk profile among patients with Lynch syndrome is continuing to evolve as more individuals are tested through multigene panel testing, with representation of larger numbers of individuals with PMS2 and MSH6 pathogenic variants compared with prior studies. In the absence of definitive risk estimates, individuals with Lynch syndrome are screened for breast cancer on the basis of family history.
Refer to the Lynch Syndrome section of the Clinical Management of Other Hereditary Breast and/or Gynecologic Cancer Syndromes section of this summary for information about clinical management of Lynch syndrome.
Li-Fraumeni Syndrome (LFS)
Breast cancer is also a component of the rare LFS, in which germline variants of the TP53 gene on chromosome 17p have been documented. Located on chromosome 17p, TP53 encodes a 53kd nuclear phosphoprotein that binds DNA sequences and functions as a negative regulator of cell growth and proliferation in the setting of DNA damage. It is also an active component of programmed cell death. Inactivation of the TP53 gene or disruption of the protein product is thought to allow the persistence of damaged DNA and the possible development of malignant cells. Widely used clinical diagnostic criteria for LFS were originally developed by Chompret et al. in 2001 (called the Chompret Criteria) and revised in 2009 based on additional emerging data.
LFS is characterized by premenopausal breast cancer in combination with childhood sarcoma, brain tumors, leukemia, and adrenocortical carcinoma.
Germline variants in TP53 are thought to account for fewer than 1% of breast cancer cases. TP53-associated breast cancer is often human epidermal growth factor receptor two (HER2/neu)–positive, in addition to being estrogen receptor (ER)–positive, progesterone receptor (PR)–positive, or both. Evidence also exists that patients treated for a TP53-related tumor with chemotherapy or radiation therapy may be at risk of a treatment-related second malignancy.
Historical criteria for defining LFS
The term LFS was used for the first time in 1982, and the following criteria, which subsequently became the classical definition of the syndrome, were proposed by Li and Fraumeni in 1988 :
- Sarcoma before age 45 years;
- A first-degree relative (FDR) with cancer before age 45 years; AND
- Another close relative (FDR or second-degree relative [SDR]) with either cancer before age 45 years or a sarcoma at any age.
Subsequently in 2001, Chompret et al. systematically developed clinical criteria for recommending TP53 genetic testing, with the narrow LFS tumor spectrum defined as sarcoma, brain tumors, breast cancer, and adrenocortical carcinoma. The criteria were as follows:
- A proband affected by a narrow-spectrum tumor before age 36 years AND at least one FDR or SDR affected by a narrow-spectrum tumor (other than breast cancer if the proband is affected by breast cancer) before age 46 years or multiple primary tumors; OR
- A proband with multiple primary tumors, two of which belong to the narrow spectrum and the first of which occurred before age 36 years, irrespective of family history; OR
- A proband with adrenocortical carcinoma irrespective of the age at onset and family history.
These criteria were revised in 2009 based on additional emerging data as follows:
- A proband with a tumor belonging to the LFS tumor spectrum* before age 46 years AND at least one FDR or SDR with an LFS tumor (except breast cancer if proband has breast cancer) before age 56 years or with multiple tumors; OR
- A proband with multiple tumors (except multiple breast tumors), two of which belong to the LFS tumor spectrum and the first of which occurred before age 46 years; OR
- A patient with adrenocortical carcinoma or choroid plexus, irrespective of family history.
*The 2009 Chompret criteria defined the LFS tumor spectrum as including the following cancers: soft tissue sarcoma, osteosarcoma, brain tumor, premenopausal breast cancer, adrenocortical carcinoma, leukemia, and lung bronchoalveolar cancer.
In 2015, Bougeard et al. revised the criteria based on data from 415 carriers of pathogenic variants, to include the presence of childhood anaplastic rhabdomyosarcoma and breast cancer before age 31 years as an indication for testing, similar to what is recommended for choroid plexus carcinoma and adrenocortical carcinoma. The criteria were revised as follows:
- A proband with a tumor belonging to the LFS tumor spectrum** before age 46 years AND at least one FDR or SDR with LFS tumor (except breast cancer if proband has breast cancer) before age 56 years or with multiple tumors; OR
- A proband with multiple tumors (except multiple breast tumors), two of which belong to the LFS tumor spectrum and the first of which occurred before age 46 years; OR
- A patient with adrenocortical carcinoma, choroid plexus tumor, or rhabdomyosarcoma of embryonal anaplastic subtype, irrespective of family history; OR
- Breast cancer before age 31 years.
**The 2015 Chompret criteria defined the LFS tumor spectrum as including the following cancers: premenopausal breast cancer, soft tissue sarcoma, osteosarcoma, central nervous system (CNS) tumor, and adrenocortical carcinoma.
Clinical characteristics of LFS
Germline TP53 pathogenic variants were identified in 17% (n = 91) of 525 samples submitted to City of Hope laboratories for clinical TP53 testing. All families with a TP53 pathogenic variant had at least one family member with a sarcoma, breast cancer, brain cancer, or adrenocortical cancer (core cancers). In addition, all eight individuals with a choroid plexus tumor had a TP53 pathogenic variant, as did 14 of the 21 individuals with childhood adrenocortical cancer. In women aged 30 to 49 years who had breast cancer but no family history of other core cancers, no TP53 variants were found.
Subsequently, a large clinical series of patients from France who were tested primarily based on the 2009 version of the Chompret criteria included 415 carriers of pathogenic variants from 214 families. In this study, 43% of carriers had multiple malignancies, and the mean age at first tumor onset was 24.9 years. The childhood tumor spectrum was characterized by osteosarcomas, adrenocortical carcinomas, CNS tumors, and soft tissue sarcomas (present in 23%–30% collectively), whereas the adult tumor spectrum primarily encompassed breast cancer (79% of females) and soft tissue sarcomas (27% of carriers). The TP53 pathogenic variant detection rate was 6% among females younger than 31 years with breast cancer and no additional features suggestive of LFS. Evaluation of genotype-phenotype correlations indicated a gradient of clinical severity, with a significantly lower mean age at onset among those with dominant-negative missense variants (21.3 years), compared with those with all types of loss-of-function variants (28.5 years) or genomic rearrangements (35.8 years). With the exception of adrenocortical carcinoma, affected children mostly harbored dominant-negative missense pathogenic variants. Among 127 female carriers of pathogenic variants with breast cancer, 31% developed contralateral breast cancer (CBC). Receptor status information was available for 40 tumors, which indicated 55% were HER2-positive, and 37% were triple-positive (i.e., ER-positive, PR-positive, and HER2-positive). There was an exceptionally high rate of multiple malignancies (43%) among carriers of pathogenic variants, of which 83% were metachronous. Treatment records were available for 64 carriers who received radiation therapy for treatment of their first tumor; of these, 19 (30%) developed 26 secondary tumors within a radiation field, with a latency of 2 to 26 years (mean, 10.7 y).
Similarly, results of 286 TP53 pathogenic variant–positive individuals in the National Cancer Institute’s LFS Study indicated a cumulative cancer incidence of almost 100% by age 70 years for both males and females. They reported substantial variations by sex, age, and cancer type. Specifically, cumulative cancer incidence reached 50% by age 31 years in females and age 46 years in males, although male risks were higher in childhood and late adulthood. Cumulative cancer incidence by sex for the top four cancers is included in Table 5. Of those with one cancer, 49% developed at least one additional cancer after a median of 10 years. Age-specific risks for developing first and second cancers were comparable.
| Cumulative Cancer Risk by Age 70 Years | ||
|---|---|---|
| aAdapted from Mai et al. | ||
| bOther cancers, such as adrenocortical carcinoma, leukemia, and lung bronchoalveolar cancer, have been considered part of the LFS cancer spectrum. | ||
| Cancer Type | Females(%) | Males (%) |
| Breast cancer | 54 | – |
| Soft tissue sarcoma | 15 | 22 |
| Brain cancer | 6 | 19 |
| Osteosarcoma | 5 | 11 |
With the increasing use of multigene (panel) tests, it is important to recognize that pathogenic variants in TP53 are unexpectedly being identified in individuals without a family history characteristic of LFS. The clinical significance of finding an isolated TP53 pathogenic variant in an individual or family who does not meet the Chompret criteria is uncertain. Consequently, it remains important to interpret cancer risks and determine optimal management strategies for individuals who are unexpectedly found to have a germline TP53 pathogenic variant, while considering their personal and family histories.
One cohort study evaluated 116 individuals with a germline TP53 pathogenic variant yearly at the National Institutes of Health Clinical Center using multimodality screening with and without gadolinium. Baseline screening identified a cancer in eight patients (6.9%) with a false-positive rate of 34.5% for MRI (n = 40). Breast cancer screening with annual breast MRI with and without contrast is recommended. Additional screening for other cancers has been studied and is evolving.
PTEN Hamartoma Tumor Syndromes (Including Cowden Syndrome)
Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome (BRRS) are part of a spectrum of conditions known collectively as PTEN hamartoma tumor syndromes (PHTS). Approximately 85% of patients diagnosed with Cowden syndrome, and approximately 60% of patients with BRRS have an identifiable PTEN pathogenic variant. In addition, PTEN pathogenic variants have been identified in patients with very diverse clinical phenotypes. The term PHTS refers to any patient with a PTEN pathogenic variant, irrespective of clinical presentation.
PTEN functions as a dual-specificity phosphatase that removes phosphate groups from tyrosine, serine, and threonine. PTEN pathogenic variants are diverse and can present as nonsense, missense, frameshift, or splice-site variants. Approximately 40% of variants are found in exon 5, which encodes the phosphatase core motif; several recurrent pathogenic variants have been observed at this location. Pathogenic variants in the 5’ end of PTEN or within the phosphatase core of PTEN tend to affect more organ systems.
Operational criteria for the diagnosis of Cowden syndrome have been published and subsequently updated. These include major, minor, and pathognomonic criteria that consist of certain mucocutaneous manifestations and adult-onset dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease). An updated set of criteria based on a systematic literature review has been suggested and is currently utilized in the National Comprehensive Cancer Network (NCCN) guidelines. Contrary to previous criteria, the authors concluded that there was insufficient evidence for any features to be classified as pathognomonic. Increased genetic testing (especially multigene panels) has identified individuals with germline PTEN pathogenic variants who do not meet diagnostic criteria for PHTS. Diagnostic criteria will need to be reconciled with these recently discovered phenotypes. Hence, it is unclear whether PHTS diagnoses should be based on clinical features or a positive PTEN genetic test result. The American College of Medical Genetics and Genomics (ACMG) suggests that referral for genetics consultation be considered for individuals with a personal history of or a first-degree relative with the following: 1) adult-onset Lhermitte-Duclos disease or 2) any three of the major or minor criteria that have been established for the diagnosis of Cowden syndrome. Detailed recommendations, including diagnostic criteria for Cowden syndrome, can be found in the NCCN and ACMG guidelines. Additionally, a predictive model that uses clinical criteria to estimate the probability of a PTEN pathogenic variant is available; a cost-effectiveness analysis suggests that germline PTEN testing is cost effective if the probability of a variant is greater than 10%.
Over a 10-year period, the International Cowden Consortium (ICC) prospectively recruited a consecutive series of adult and pediatric patients meeting relaxed ICC criteria for PTEN testing in the United States, Europe, and Asia. Most individuals did not meet the clinical criteria for a diagnosis of Cowden syndrome or BRRS. Of the 3,399 individuals recruited and tested, 295 probands (8.8%) and an additional 73 family members carried a germline PTEN pathogenic variant. The authors concluded that melanoma, kidney cancer, and colorectal cancer should be added to the spectrum of cancers associated with PTEN germline pathogenic variants (in addition to breast cancer, thyroid cancer, and endometrial cancer). This conclusion was based on the high melanoma, kidney, and colorectal cancer lifetime risk estimates found in individuals with PTEN pathogenic variants. A second study of approximately 100 patients with a germline PTEN pathogenic variant confirmed these findings and suggested a cumulative cancer risk of 85% by age 70 years.
Although PTEN pathogenic variants, which are estimated to occur in 1 in 200,000 individuals, account for a small fraction of hereditary breast cancer, the characterization of PTEN function will provide valuable insights into the signal pathway and the maintenance of normal cell physiology. Lifetime breast cancer risk is estimated to be between 25% and 50% among women with Cowden syndrome. Other studies have reported risks as high as 85%; however, there are concerns regarding selection bias in these studies. As in other forms of hereditary breast cancer, onset is often at a young age and may be bilateral. Lifetime risk of endometrial cancer is estimated to be between 19% and 28%, depending on the cohort studied, with an increased risk of premenopausal onset. Because of the low prevalence of PTEN pathogenic variants in the population, the proportion of endometrial cancer attributable to Cowden syndrome is small. There are no data that link PTEN pathogenic variants to an increased risk of ovarian cancer. Skin manifestations include multiple trichilemmomas, oral fibromas and papillomas, and acral, palmar, and plantar keratoses. History or observation of the characteristic skin features raises a suspicion of Cowden syndrome. CNS manifestations include macrocephaly, developmental delay, and dysplastic gangliocytomas of the cerebellum. (Refer to the PDQ summaries on Genetics of Colorectal Cancer and Genetics of Skin Cancer for more information about PTEN hamartoma tumor syndromes [including Cowden syndrome].)
Hereditary Diffuse Gastric Cancer (HDGC)
For more information about HDGC, see the following:
- For general information about HDGC, see Hereditary Diffuse Gastric Cancer.
- For information about HDGC-associated lobular breast cancer, see the Breast Cancer Risk section in Hereditary Diffuse Gastric Cancer.
- For information about management of breast cancer risk in HDGC, see the Breast Cancer Risk Management section in Hereditary Diffuse Gastric Cancer.
Peutz-Jeghers Syndrome (PJS)
PJS is an early-onset autosomal dominant disorder characterized by melanocytic macules on the lips, the perioral region, and buccal region; and multiple gastrointestinal polyps, both hamartomatous and adenomatous. Germline pathogenic variants in the STK11 gene at chromosome 19p13.3 have been identified in the vast majority of PJS families. The most common cancers in PJS are gastrointestinal. However, other organs are at increased risk of developing malignancies. For example, the cumulative risks have been estimated to be 32% to 54% for breast cancer and 21% for ovarian cancer (mainly ovarian sex-cord tumors). The risk for pancreatic cancer has been estimated to be more than 100-fold higher than that in the general population. A systematic review found a lifetime cumulative cancer risk, all sites combined, of up to 93% in patients with PJS. Table 6 shows the cumulative risk of these tumors.
Females with PJS are also predisposed to the development of cervical adenoma malignum, a rare and very aggressive adenocarcinoma of the cervix. In addition, females with PJS commonly develop benign ovarian sex-cord tumors with annular tubules, whereas males with PJS are predisposed to development of Sertoli-cell testicular tumors; although neither of these two tumor types is malignant, they can cause symptoms related to increased estrogen production.
Although the risk of malignancy appears to be exceedingly high in individuals with PJS based on the published literature, the possibility that selection and referral biases have resulted in overestimates of these risks should be considered.
| Site | Age (y) | Cumulative Risk (%)b | Reference(s) |
|---|---|---|---|
| GI = gastrointestinal. | |||
| aReprinted with permission from Macmillan Publishers Ltd: Gastroenterology , copyright 2010. | |||
| bAll cumulative risks were increased compared with the general population (P< .05), with the exception of cervix and testes. | |||
| cGI cancers include colorectal, small intestinal, gastric, esophageal, and pancreatic. | |||
| dWesterman et al.: GI cancer does not include pancreatic cancer. | |||
| eDid not include adenoma malignum of the cervix or Sertoli cell tumors of the testes. | |||
| Any cancer | 60–70 | 37–93 | |
| GI cancerc,d | 60–70 | 38–66 | |
| Gynecological cancer | 60–70 | 13–18 | |
| Per origin | |||
| Stomach | 65 | 29 | |
| Small bowel | 65 | 13 | |
| Colorectum | 65 | 39 | |
| Pancreas | 65–70 | 11–36 | |
| Lung | 65–70 | 7–17 | |
| Breast | 60–70 | 32–54 | |
| Uterus | 65 | 9 | |
| Ovary | 65 | 21 | |
| Cervixe | 65 | 10 | |
| Testese | 65 | 9 | |
Peutz-Jeghers gene(s)
PJS is caused by pathogenic variants in the STK11 (also called LKB1) tumor suppressor gene located on chromosome 19p13. Unlike the adenomas seen in familial adenomatous polyposis, the polyps arising in PJS are hamartomas. Studies of the hamartomatous polyps and cancers of PJS show allelic imbalance (LOH) consistent with the two-hit hypothesis, demonstrating that STK11 is a tumor suppressor gene. However, heterozygous STK11 knockout mice develop hamartomas without inactivation of the remaining wild-type allele, suggesting that haploinsufficiency may be sufficient for initial tumor development in PJS. Subsequently, the cancers that develop in STK11 +/- mice do show LOH; indeed, compound mutant mice heterozygous for pathogenic variants in STK11 +/- and homozygous for pathogenic variants in TP53 -/- have accelerated development of both hamartomas and cancers.
Germline variants of the STK11 gene represent a spectrum of nonsense, frameshift, and missense variants, and splice-site variants and large deletions.
Approximately 85% of variants are localized to regions of the kinase domain of the expressed protein. No strong genotype-phenotype correlations have been identified. Up to 30% of variants are large deletions involving one or more exons of STK11, underscoring the importance of deletion analysis in suspected cases of PJS.
STK11 has been unequivocally demonstrated to cause PJS. Although earlier estimates using direct DNA sequencing showed a 50% pathogenic variant detection rate in STK11, studies adding techniques to detect large deletions have found pathogenic variants in up to 94% of individuals meeting clinical criteria for PJS. Given the results of these studies, it is unlikely that other major genes cause PJS.
Clinical management
The high cumulative risk of cancers in PJS has led to the various screening recommendations summarized in the table of Published Recommendations for Diagnosis and Surveillance of Peutz-Jeghers Syndrome (PJS) in the PDQ summary on Genetics of Colorectal Cancer.
PALB2
PALB2 (partner and localizer of BRCA2) interacts with the BRCA2 protein and plays a role in homologous recombination and double-stranded DNA repair. Similar to BRIP1 and BRCA2, biallelic pathogenic variants in PALB2 have also been shown to cause Fanconi anemia.
PALB2 pathogenic variants have been screened for in multiple small studies of familial and early-onset breast cancer in multiple populations. Pathogenic variant prevalence has ranged from 0.4% to 3.9%. Similar to BRIP1 and CHEK2, there was incomplete segregation of PALB2 pathogenic variants in families with hereditary breast cancer. Among 559 cases with CBC and 565 matched controls with unilateral breast cancer, pathogenic (truncating) PALB2 pathogenic variants were identified in 0.9% of cases and in none of the controls (RR, 5.3; 95% CI, 1.8–13.2).
Data based on 154 families with loss-of-function PALB2 variants suggest that this gene may be an important cause of hereditary breast cancer, with risks that overlap with BRCA2. In this study, analysis of 362 family members from 154 families with PALB2 pathogenic variants indicated that the AR of female breast cancer by age 70 years ranged from 33% (95% CI, 24%–44%) for those with no family history of breast cancer to 58% (95% CI, 50%–66%) for those with two or more FDRs with early-onset breast cancer. Furthermore, among 63 breast cancer cases in which HER2 status was known, 30% had triple-negative breast cancer (TNBC). An earlier Finnish study reported on a PALB2 founder pathogenic variant (c.1592delT) that confers a 40% risk of breast cancer to age 70 years and is associated with a high incidence (54%) of TNBC and lower survival. Pathogenic variants have been observed in early-onset and familial breast cancer in many populations. A large report of 1,824 patients with TNBC unselected for family history, recruited through 12 studies, identified 1.2% with a PALB2 pathogenic variant. (Refer to the BRCA2-associated breast cancer pathology section in BRCA1 and BRCA2: Cancer Risks and Management for more information about this study.)
In a later Polish study of more than 12,529 unselected women with breast cancer and 4,702 controls, PALB2 pathogenic variants were detected in 116 cases (0.93%; 95% CI, 0.76%–1.09%) and 10 controls (0.21%; 95% CI, 0.08%–0.34%), with an OR for breast cancer of 4.39 (95% CI, 2.30–8.37). The study findings confirm a substantially elevated risk of breast cancer (24%–40%) among women with a PALB2 pathogenic variant up to age 75 years. The 5-year cumulative incidence of CBC was 10% among those with a PALB2 pathogenic variant, compared with 17% among those with a BRCA1 pathogenic variant and 3% among those without a variant in either gene. Furthermore, the 10-year survival for women with a PALB2 pathogenic variant and breast cancer was 48% (95% CI, 36.5%–63.2%), compared with 72.0% among those with a BRCA1 pathogenic variant and 74.7% among those without a variant in either gene. Among PALB2 carriers, breast tumors 2 cm or larger had substantially worse outcomes (32.4% 10-year survival), compared with tumors smaller than 2 cm (82.4% 10-year survival). Approximately one-third of those with a PALB2 pathogenic variant had TNBC, and the average age at breast cancer diagnosis was 53.3 years.
A similar case-control study from China enrolled 16,501 unselected patients with breast cancer and 5,890 controls. These patients were screened for PALB2, BRCA1, and BRCA2 pathogenic variants. The prevalence of PALB2 pathogenic variants was 0.97% in the cases and 0.19% in the controls. The OR for breast cancer for carriers was 5.23 (95% CI, 2.84–9.65; P > .0001). PALB2 carriers were more likely to be age 30 years or younger (6.88% vs. 3.56%; P = .04). PALB2 carriers were also more likely to have TNBCs (22.83% vs. 13.56%; P = .004), larger tumors (tumor size ≥2 cm, 55.93% vs. 45.93%; P = .04), node-positive tumors (49.60% vs. 38.80%; P = .018), and CBCs (6.29% vs. 2.01%; P = .003). Additionally, PALB2 carriers were more likely to have a family history of breast and/or ovarian cancers (20.63% vs. 7.96%; P< .0001).
In the largest study to date, 524 families with pathogenic variants in PALB2 recruited through an international effort, the RR of breast cancer in women was reported to be 7.18 (95% CI, 5.82–8.85), with a 53% risk (95% CI, 44%–63%) of breast cancer to age 80 years. Additional elevated risks reported included ovarian cancer (RR, 2.91; 95% CI, 1.40–6.04) with a lifetime risk of 5% (95% CI, 2%–10%), pancreatic cancer (RR, 2.37; 95% CI, 1.24–4.50) with a lifetime risk of 2% to 3% (95% CI for women, 1%–4%; 95% CI for men, 2%–5%), and male breast cancer (RR, 7.34; 95% CI, 1.28–42.18) with a lifetime risk of 1% (95% CI, 0.2%–5%). These findings confirm the role of PALB2 as an inherited breast cancer gene in women, while firmly establishing an association with ovarian, pancreatic, and male breast cancers.
Male breast cancer has been observed in PALB2 pathogenic variant–positive breast cancer families. In a study of 115 male breast cancer cases in which 18 men had BRCA2 pathogenic variants, an additional two men had either a pathogenic or predicted pathogenic PALB2 variant (accounting for about 10% of germline variants in the study and 1%–2% of the total sample). The RR of breast cancer for male carriers of PALB2 pathogenic variants compared with that seen in the general population was estimated to be 8.30 (95% CI, 0.77–88.56; P = .08) in the study of 154 families.
After the identification of PALB2 pathogenic variants in pancreatic tumors and the detection of germline pathogenic variants in 3% of 96 familial pancreatic patients, numerous studies have pointed to a role for PALB2 in pancreatic cancer. PALB2 pathogenic variants were detected in 3.7% of 81 familial pancreatic cancer families and in 2.1% of 94 BRCA1/BRCA2 pathogenic variant–negative breast cancer patients who had either a personal or family history of pancreatic cancer. Two relatively small studies—one of 77 BRCA1/BRCA2 pathogenic variant–negative probands with a personal or family history of pancreatic cancer, one-half of whom were of AJ descent, and another study of 29 Italian pancreatic cancer patients with a personal or family history of breast or ovarian cancer—failed to detect any PALB2 pathogenic variants. A sixfold increase in pancreatic cancer was observed in the relatives of 33 BRCA1/BRCA2-negative, PALB2 pathogenic variant–positive breast cancer probands.
Overall, the observed prevalence of PALB2 pathogenic variants in familial breast cancer varied depending on ascertainment relative to personal and family history of pancreatic and ovarian cancers, but in all studies, the observed pathogenic variant rate was lower than 4%. Data suggest that the RR of breast cancer may overlap with that of BRCA2, particularly in those with a strong family history; thus, it remains important to refine cancer risk estimates in larger studies. Furthermore, the risk of other cancers (e.g., pancreatic) is poorly defined. Given the low PALB2 pathogenic variant prevalence in the population, additional data are needed to define best candidates for testing and appropriate management.
De Novo Pathogenic Variant Rate
Until the 1990s, the diagnosis of genetically inherited breast and ovarian cancer syndromes was based on clinical manifestations and family history. Now that some of the genes involved in these syndromes have been identified, a few studies have attempted to estimate the spontaneous pathogenic variant rate (de novo pathogenic variant rate) in these populations. Interestingly, PJS, PTEN hamartoma syndromes, and LFS are all thought to have high rates of spontaneous pathogenic variants, in the 10% to 30% range, while estimates of de novo pathogenic variants in the BRCA genes are thought to be low, primarily on the basis of the few case reports published. Additionally, there has been only one case series of breast cancer patients who were tested for BRCA pathogenic variants in which a de novo variant was identified. Specifically, in this study of 193 patients with sporadic breast cancer, 17 pathogenic variants were detected, one of which was confirmed to be a de novo pathogenic variant. As such, the de novo pathogenic variant rate appears to be low and fall into the 5% or less range, based on the limited studies performed. Similarly, estimates of de novo pathogenic variants in the MMR genes associated with Lynch syndrome are thought to be low, in the 0.9% to 5% range. However, these estimates of spontaneous pathogenic variant rates in the BRCA genes and Lynch syndrome genes seem to overlap with the estimates of nonpaternity rates in various populations (0.6%–3.3%), making the de novo pathogenic variant rate for these genes relatively low.
References
- Vasen HF: Clinical description of the Lynch syndrome [hereditary nonpolyposis colorectal cancer (HNPCC)]. Fam Cancer 4 (3): 219-25, 2005.
- Jascur T, Boland CR: Structure and function of the components of the human DNA mismatch repair system. Int J Cancer 119 (9): 2030-5, 2006.
- Papadopoulos N, Nicolaides NC, Wei YF, et al.: Mutation of a mutL homolog in hereditary colon cancer. Science 263 (5153): 1625-9, 1994.
- Peltomäki P, Vasen HF: Mutations predisposing to hereditary nonpolyposis colorectal cancer: database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 113 (4): 1146-58, 1997.
- Akiyama Y, Sato H, Yamada T, et al.: Germ-line mutation of the hMSH6/GTBP gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Cancer Res 57 (18): 3920-3, 1997.
- Miyaki M, Konishi M, Tanaka K, et al.: Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet 17 (3): 271-2, 1997.
- Huang J, Kuismanen SA, Liu T, et al.: MSH6 and MSH3 are rarely involved in genetic predisposition to nonpolypotic colon cancer. Cancer Res 61 (4): 1619-23, 2001.
- Nicolaides NC, Papadopoulos N, Liu B, et al.: Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 371 (6492): 75-80, 1994.
- Hendriks YM, Jagmohan-Changur S, van der Klift HM, et al.: Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (Lynch syndrome). Gastroenterology 130 (2): 312-22, 2006.
- Worthley DL, Walsh MD, Barker M, et al.: Familial mutations in PMS2 can cause autosomal dominant hereditary nonpolyposis colorectal cancer. Gastroenterology 128 (5): 1431-6, 2005.
- Vasen HF, Mecklin JP, Khan PM, et al.: The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 34 (5): 424-5, 1991.
- Vasen HF, Watson P, Mecklin JP, et al.: New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 116 (6): 1453-6, 1999.
- Rodriguez-Bigas MA, Boland CR, Hamilton SR, et al.: A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst 89 (23): 1758-62, 1997.
- Umar A, Boland CR, Terdiman JP, et al.: Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 96 (4): 261-8, 2004.
- Watson P, Lynch HT: Cancer risk in mismatch repair gene mutation carriers. Fam Cancer 1 (1): 57-60, 2001.
- Vasen HF, Wijnen JT, Menko FH, et al.: Cancer risk in families with hereditary nonpolyposis colorectal cancer diagnosed by mutation analysis. Gastroenterology 110 (4): 1020-7, 1996.
- Aarnio M, Mecklin JP, Aaltonen LA, et al.: Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer 64 (6): 430-3, 1995.
- Watson P, Lynch HT: Extracolonic cancer in hereditary nonpolyposis colorectal cancer. Cancer 71 (3): 677-85, 1993.
- Brown GJ, St John DJ, Macrae FA, et al.: Cancer risk in young women at risk of hereditary nonpolyposis colorectal cancer: implications for gynecologic surveillance. Gynecol Oncol 80 (3): 346-9, 2001.
- Aarnio M, Sankila R, Pukkala E, et al.: Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer 81 (2): 214-8, 1999.
- Ten Broeke SW, van der Klift HM, Tops CMJ, et al.: Cancer Risks for PMS2-Associated Lynch Syndrome. J Clin Oncol 36 (29): 2961-2968, 2018.
- Møller P, Seppälä TT, Bernstein I, et al.: Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database. Gut 67 (7): 1306-1316, 2018.
- Grindedal EM, Renkonen-Sinisalo L, Vasen H, et al.: Survival in women with MMR mutations and ovarian cancer: a multicentre study in Lynch syndrome kindreds. J Med Genet 47 (2): 99-102, 2010.
- Pal T, Permuth-Wey J, Sellers TA: A review of the clinical relevance of mismatch-repair deficiency in ovarian cancer. Cancer 113 (4): 733-42, 2008.
- Jensen UB, Sunde L, Timshel S, et al.: Mismatch repair defective breast cancer in the hereditary nonpolyposis colorectal cancer syndrome. Breast Cancer Res Treat 120 (3): 777-82, 2010.
- Shanley S, Fung C, Milliken J, et al.: Breast cancer immunohistochemistry can be useful in triage of some HNPCC families. Fam Cancer 8 (3): 251-5, 2009.
- Walsh MD, Buchanan DD, Cummings MC, et al.: Lynch syndrome-associated breast cancers: clinicopathologic characteristics of a case series from the colon cancer family registry. Clin Cancer Res 16 (7): 2214-24, 2010.
- Buerki N, Gautier L, Kovac M, et al.: Evidence for breast cancer as an integral part of Lynch syndrome. Genes Chromosomes Cancer 51 (1): 83-91, 2012.
- Win AK, Young JP, Lindor NM, et al.: Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol 30 (9): 958-64, 2012.
- Win AK, Lindor NM, Young JP, et al.: Risks of primary extracolonic cancers following colorectal cancer in lynch syndrome. J Natl Cancer Inst 104 (18): 1363-72, 2012.
- Harkness EF, Barrow E, Newton K, et al.: Lynch syndrome caused by MLH1 mutations is associated with an increased risk of breast cancer: a cohort study. J Med Genet 52 (8): 553-6, 2015.
- Win AK, Lindor NM, Jenkins MA: Risk of breast cancer in Lynch syndrome: a systematic review. Breast Cancer Res 15 (2): R27, 2013.
- Goldberg M, Bell K, Aronson M, et al.: Association between the Lynch syndrome gene MSH2 and breast cancer susceptibility in a Canadian familial cancer registry. J Med Genet 54 (11): 742-746, 2017.
- Roberts ME, Jackson SA, Susswein LR, et al.: MSH6 and PMS2 germ-line pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genet Med 20 (10): 1167-1174, 2018.
- Espenschied CR, LaDuca H, Li S, et al.: Multigene Panel Testing Provides a New Perspective on Lynch Syndrome. J Clin Oncol 35 (22): 2568-2575, 2017.
- Lu HM, Li S, Black MH, et al.: Association of Breast and Ovarian Cancers With Predisposition Genes Identified by Large-Scale Sequencing. JAMA Oncol 5 (1): 51-57, 2019.
- Latham A, Srinivasan P, Kemel Y, et al.: Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J Clin Oncol 37 (4): 286-295, 2019.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 1.2021. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2020. Available online with free registration. Last accessed November 8, 2023.
- Harris CC, Hollstein M: Clinical implications of the p53 tumor-suppressor gene. N Engl J Med 329 (18): 1318-27, 1993.
- Malkin D: The Li-Fraumeni syndrome. Cancer: Principles and Practice of Oncology Updates 7(7): 1-14, 1993.
- Gonzalez KD, Noltner KA, Buzin CH, et al.: Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 27 (8): 1250-6, 2009.
- Chompret A, Abel A, Stoppa-Lyonnet D, et al.: Sensitivity and predictive value of criteria for p53 germline mutation screening. J Med Genet 38 (1): 43-7, 2001.
- Tinat J, Bougeard G, Baert-Desurmont S, et al.: 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol 27 (26): e108-9; author reply e110, 2009.
- Bottomley RH, Condit PT: Cancer families. Cancer Bull 20: 22-24, 1968.
- Bougeard G, Renaux-Petel M, Flaman JM, et al.: Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J Clin Oncol 33 (21): 2345-52, 2015.
- Sidransky D, Tokino T, Helzlsouer K, et al.: Inherited p53 gene mutations in breast cancer. Cancer Res 52 (10): 2984-6, 1992.
- Wilson JR, Bateman AC, Hanson H, et al.: A novel HER2-positive breast cancer phenotype arising from germline TP53 mutations. J Med Genet 47 (11): 771-4, 2010.
- Melhem-Bertrandt A, Bojadzieva J, Ready KJ, et al.: Early onset HER2-positive breast cancer is associated with germline TP53 mutations. Cancer 118 (4): 908-13, 2012.
- Masciari S, Dillon DA, Rath M, et al.: Breast cancer phenotype in women with TP53 germline mutations: a Li-Fraumeni syndrome consortium effort. Breast Cancer Res Treat 133 (3): 1125-30, 2012.
- Pearson AD, Craft AW, Ratcliffe JM, et al.: Two families with the Li-Fraumeni cancer family syndrome. J Med Genet 19 (5): 362-5, 1982.
- Li FP, Fraumeni JF, Mulvihill JJ, et al.: A cancer family syndrome in twenty-four kindreds. Cancer Res 48 (18): 5358-62, 1988.
- Bougeard G, Sesboüé R, Baert-Desurmont S, et al.: Molecular basis of the Li-Fraumeni syndrome: an update from the French LFS families. J Med Genet 45 (8): 535-8, 2008.
- Mai PL, Best AF, Peters JA, et al.: Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 122 (23): 3673-3681, 2016.
- Kamihara J, Rana HQ, Garber JE: Germline TP53 mutations and the changing landscape of Li-Fraumeni syndrome. Hum Mutat 35 (6): 654-62, 2014.
- Mai PL, Khincha PP, Loud JT, et al.: Prevalence of Cancer at Baseline Screening in the National Cancer Institute Li-Fraumeni Syndrome Cohort. JAMA Oncol 3 (12): 1640-1645, 2017.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 2.2024. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed October 31, 2023.
- Villani A, Tabori U, Schiffman J, et al.: Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. Lancet Oncol 12 (6): 559-67, 2011.
- Masciari S, Van den Abbeele AD, Diller LR, et al.: F18-fluorodeoxyglucose-positron emission tomography/computed tomography screening in Li-Fraumeni syndrome. JAMA 299 (11): 1315-9, 2008.
- Zhou XP, Waite KA, Pilarski R, et al.: Germline PTEN promoter mutations and deletions in Cowden/Bannayan-Riley-Ruvalcaba syndrome result in aberrant PTEN protein and dysregulation of the phosphoinositol-3-kinase/Akt pathway. Am J Hum Genet 73 (2): 404-11, 2003.
- Mester J, Eng C: When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet 163C (2): 114-21, 2013.
- Eng C: PTEN: one gene, many syndromes. Hum Mutat 22 (3): 183-98, 2003.
- Marsh DJ, Kum JB, Lunetta KL, et al.: PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet 8 (8): 1461-72, 1999.
- Pilarski R, Eng C: Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet 41 (5): 323-6, 2004.
- Eng C: PTEN Hamartoma Tumor Syndrome (PHTS). In: Adam MP, Feldman J, Mirzaa GM, et al., eds.: GeneReviews. University of Washington, Seattle, 1993-2024, pp. Available online. Last accessed March 6, 2024.
- Pilarski R, Burt R, Kohlman W, et al.: Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst 105 (21): 1607-16, 2013.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 2.2022. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2022. Available online with free registration. Last accessed August 3, 2023.
- Hampel H, Bennett RL, Buchanan A, et al.: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 17 (1): 70-87, 2015.
- Ngeow J, Liu C, Zhou K, et al.: Detecting Germline PTEN Mutations Among At-Risk Patients With Cancer: An Age- and Sex-Specific Cost-Effectiveness Analysis. J Clin Oncol 33 (23): 2537-44, 2015.
- Tan MH, Mester JL, Ngeow J, et al.: Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 18 (2): 400-7, 2012.
- Bubien V, Bonnet F, Brouste V, et al.: High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet 50 (4): 255-63, 2013.
- Myers MP, Tonks NK: PTEN: sometimes taking it off can be better than putting it on. Am J Hum Genet 61 (6): 1234-8, 1997.
- Hobert JA, Eng C: PTEN hamartoma tumor syndrome: an overview. Genet Med 11 (10): 687-94, 2009.
- Nieuwenhuis MH, Kets CM, Murphy-Ryan M, et al.: Cancer risk and genotype-phenotype correlations in PTEN hamartoma tumor syndrome. Fam Cancer 13 (1): 57-63, 2014.
- Ngeow J, Stanuch K, Mester JL, et al.: Second malignant neoplasms in patients with Cowden syndrome with underlying germline PTEN mutations. J Clin Oncol 32 (17): 1818-24, 2014.
- Olopade OI, Weber BL: Breast cancer genetics: toward molecular characterization of individuals at increased risk for breast cancer: part I. Cancer: Principles and Practice of Oncology Updates 12 (10): 1-12, 1998.
- Pilarski R, Stephens JA, Noss R, et al.: Predicting PTEN mutations: an evaluation of Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome clinical features. J Med Genet 48 (8): 505-12, 2011.
- Nelen MR, Padberg GW, Peeters EA, et al.: Localization of the gene for Cowden disease to chromosome 10q22-23. Nat Genet 13 (1): 114-6, 1996.
- Lachlan KL, Lucassen AM, Bunyan D, et al.: Cowden syndrome and Bannayan Riley Ruvalcaba syndrome represent one condition with variable expression and age-related penetrance: results of a clinical study of PTEN mutation carriers. J Med Genet 44 (9): 579-85, 2007.
- Peutz JL: Very remarkable case of familial polyposis of mucous membrane of intestinal tract and nasopharynx accompanied by peculiar pigmentations of skin and mucous membrane. Ned Tijdschr Geneeskd 10: 134-146, 1921.
- Jeghers H, McKusick VA, Katz KH: Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med 241 (26): 1031-6, 1949.
- Spigelman AD, Murday V, Phillips RK: Cancer and the Peutz-Jeghers syndrome. Gut 30 (11): 1588-90, 1989.
- Aretz S, Stienen D, Uhlhaas S, et al.: High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum Mutat 26 (6): 513-9, 2005.
- Hemminki A, Markie D, Tomlinson I, et al.: A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 391 (6663): 184-7, 1998.
- Jenne DE, Reimann H, Nezu J, et al.: Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 18 (1): 38-43, 1998.
- Boudeau J, Kieloch A, Alessi DR, et al.: Functional analysis of LKB1/STK11 mutants and two aberrant isoforms found in Peutz-Jeghers Syndrome patients. Hum Mutat 21 (2): 172, 2003.
- Lim W, Hearle N, Shah B, et al.: Further observations on LKB1/STK11 status and cancer risk in Peutz-Jeghers syndrome. Br J Cancer 89 (2): 308-13, 2003.
- Giardiello FM, Brensinger JD, Tersmette AC, et al.: Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 119 (6): 1447-53, 2000.
- Hearle N, Schumacher V, Menko FH, et al.: Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 12 (10): 3209-15, 2006.
- Lim W, Olschwang S, Keller JJ, et al.: Relative frequency and morphology of cancers in STK11 mutation carriers. Gastroenterology 126 (7): 1788-94, 2004.
- van Lier MG, Wagner A, Mathus-Vliegen EM, et al.: High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol 105 (6): 1258-64; author reply 1265, 2010.
- Srivatsa PJ, Keeney GL, Podratz KC: Disseminated cervical adenoma malignum and bilateral ovarian sex cord tumors with annular tubules associated with Peutz-Jeghers syndrome. Gynecol Oncol 53 (2): 256-64, 1994.
- Scully RE: Sex cord tumor with annular tubules a distinctive ovarian tumor of the Peutz-Jeghers syndrome. Cancer 25 (5): 1107-21, 1970.
- Westerman AM, Entius MM, de Baar E, et al.: Peutz-Jeghers syndrome: 78-year follow-up of the original family. Lancet 353 (9160): 1211-5, 1999.
- Mehenni H, Resta N, Park JG, et al.: Cancer risks in LKB1 germline mutation carriers. Gut 55 (7): 984-90, 2006.
- Gruber SB, Entius MM, Petersen GM, et al.: Pathogenesis of adenocarcinoma in Peutz-Jeghers syndrome. Cancer Res 58 (23): 5267-70, 1998.
- Wang ZJ, Ellis I, Zauber P, et al.: Allelic imbalance at the LKB1 (STK11) locus in tumours from patients with Peutz-Jeghers' syndrome provides evidence for a hamartoma-(adenoma)-carcinoma sequence. J Pathol 188 (1): 9-13, 1999.
- Miyoshi H, Nakau M, Ishikawa TO, et al.: Gastrointestinal hamartomatous polyposis in Lkb1 heterozygous knockout mice. Cancer Res 62 (8): 2261-6, 2002.
- Nakau M, Miyoshi H, Seldin MF, et al.: Hepatocellular carcinoma caused by loss of heterozygosity in Lkb1 gene knockout mice. Cancer Res 62 (16): 4549-53, 2002.
- Takeda H, Miyoshi H, Kojima Y, et al.: Accelerated onsets of gastric hamartomas and hepatic adenomas/carcinomas in Lkb1+/-p53-/- compound mutant mice. Oncogene 25 (12): 1816-20, 2006.
- Amos CI, Keitheri-Cheteri MB, Sabripour M, et al.: Genotype-phenotype correlations in Peutz-Jeghers syndrome. J Med Genet 41 (5): 327-33, 2004.
- Reid S, Schindler D, Hanenberg H, et al.: Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet 39 (2): 162-4, 2007.
- Ding YC, Steele L, Kuan CJ, et al.: Mutations in BRCA2 and PALB2 in male breast cancer cases from the United States. Breast Cancer Res Treat 126 (3): 771-8, 2011.
- Rahman N, Seal S, Thompson D, et al.: PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet 39 (2): 165-7, 2007.
- Erkko H, Dowty JG, Nikkilä J, et al.: Penetrance analysis of the PALB2 c.1592delT founder mutation. Clin Cancer Res 14 (14): 4667-71, 2008.
- Heikkinen T, Kärkkäinen H, Aaltonen K, et al.: The breast cancer susceptibility mutation PALB2 1592delT is associated with an aggressive tumor phenotype. Clin Cancer Res 15 (9): 3214-22, 2009.
- Ding YC, Steele L, Chu LH, et al.: Germline mutations in PALB2 in African-American breast cancer cases. Breast Cancer Res Treat 126 (1): 227-30, 2011.
- Foulkes WD, Ghadirian P, Akbari MR, et al.: Identification of a novel truncating PALB2 mutation and analysis of its contribution to early-onset breast cancer in French-Canadian women. Breast Cancer Res 9 (6): R83, 2007.
- Casadei S, Norquist BM, Walsh T, et al.: Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res 71 (6): 2222-9, 2011.
- Southey MC, Teo ZL, Dowty JG, et al.: A PALB2 mutation associated with high risk of breast cancer. Breast Cancer Res 12 (6): R109, 2010.
- Hellebrand H, Sutter C, Honisch E, et al.: Germline mutations in the PALB2 gene are population specific and occur with low frequencies in familial breast cancer. Hum Mutat 32 (6): E2176-88, 2011.
- Bogdanova N, Sokolenko AP, Iyevleva AG, et al.: PALB2 mutations in German and Russian patients with bilateral breast cancer. Breast Cancer Res Treat 126 (2): 545-50, 2011.
- Wong MW, Nordfors C, Mossman D, et al.: BRIP1, PALB2, and RAD51C mutation analysis reveals their relative importance as genetic susceptibility factors for breast cancer. Breast Cancer Res Treat 127 (3): 853-9, 2011.
- Zheng Y, Zhang J, Niu Q, et al.: Novel germline PALB2 truncating mutations in African American breast cancer patients. Cancer 118 (5): 1362-70, 2012.
- Tischkowitz M, Capanu M, Sabbaghian N, et al.: Rare germline mutations in PALB2 and breast cancer risk: a population-based study. Hum Mutat 33 (4): 674-80, 2012.
- Fernandes PH, Saam J, Peterson J, et al.: Comprehensive sequencing of PALB2 in patients with breast cancer suggests PALB2 mutations explain a subset of hereditary breast cancer. Cancer 120 (7): 963-7, 2014.
- Janatova M, Kleibl Z, Stribrna J, et al.: The PALB2 gene is a strong candidate for clinical testing in BRCA1- and BRCA2-negative hereditary breast cancer. Cancer Epidemiol Biomarkers Prev 22 (12): 2323-32, 2013.
- Teo ZL, Sawyer SD, James PA, et al.: The incidence of PALB2 c.3113G>A in women with a strong family history of breast and ovarian cancer attending familial cancer centres in Australia. Fam Cancer 12 (4): 587-95, 2013.
- Snyder C, Metcalfe K, Sopik V, et al.: Prevalence of PALB2 mutations in the Creighton University Breast Cancer Family Registry. Breast Cancer Res Treat 150 (3): 637-41, 2015.
- Nguyen-Dumont T, Hammet F, Mahmoodi M, et al.: Mutation screening of PALB2 in clinically ascertained families from the Breast Cancer Family Registry. Breast Cancer Res Treat 149 (2): 547-54, 2015.
- Damiola F, Schultz I, Barjhoux L, et al.: Mutation analysis of PALB2 gene in French breast cancer families. Breast Cancer Res Treat 154 (3): 463-71, 2015.
- Antoniou AC, Casadei S, Heikkinen T, et al.: Breast-cancer risk in families with mutations in PALB2. N Engl J Med 371 (6): 497-506, 2014.
- Couch FJ, Hart SN, Sharma P, et al.: Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol 33 (4): 304-11, 2015.
- Cybulski C, Kluźniak W, Huzarski T, et al.: Clinical outcomes in women with breast cancer and a PALB2 mutation: a prospective cohort analysis. Lancet Oncol 16 (6): 638-44, 2015.
- Zhou J, Wang H, Fu F, et al.: Spectrum of PALB2 germline mutations and characteristics of PALB2-related breast cancer: Screening of 16,501 unselected patients with breast cancer and 5890 controls by next-generation sequencing. Cancer 126 (14): 3202-3208, 2020.
- Yang X, Leslie G, Doroszuk A, et al.: Cancer Risks Associated With Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J Clin Oncol 38 (7): 674-685, 2020.
- Jones S, Hruban RH, Kamiyama M, et al.: Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 324 (5924): 217, 2009.
- Slater EP, Langer P, Niemczyk E, et al.: PALB2 mutations in European familial pancreatic cancer families. Clin Genet 78 (5): 490-4, 2010.
- Hofstatter EW, Domchek SM, Miron A, et al.: PALB2 mutations in familial breast and pancreatic cancer. Fam Cancer 10 (2): 225-31, 2011.
- Stadler ZK, Salo-Mullen E, Sabbaghian N, et al.: Germline PALB2 mutation analysis in breast-pancreas cancer families. J Med Genet 48 (8): 523-5, 2011.
- Ghiorzo P, Pensotti V, Fornarini G, et al.: Contribution of germline mutations in the BRCA and PALB2 genes to pancreatic cancer in Italy. Fam Cancer 11 (1): 41-7, 2012.
- Westerman AM, Entius MM, Boor PP, et al.: Novel mutations in the LKB1/STK11 gene in Dutch Peutz-Jeghers families. Hum Mutat 13 (6): 476-81, 1999.
- Schreibman IR, Baker M, Amos C, et al.: The hamartomatous polyposis syndromes: a clinical and molecular review. Am J Gastroenterol 100 (2): 476-90, 2005.
- Gonzalez KD, Buzin CH, Noltner KA, et al.: High frequency of de novo mutations in Li-Fraumeni syndrome. J Med Genet 46 (10): 689-93, 2009.
- Bendig I, Mohr N, Kramer F, et al.: Identification of novel TP53 mutations in familial and sporadic cancer cases of German and Swiss origin. Cancer Genet Cytogenet 154 (1): 22-6, 2004.
- De Leeneer K, Coene I, Crombez B, et al.: Prevalence of BRCA1/2 mutations in sporadic breast/ovarian cancer patients and identification of a novel de novo BRCA1 mutation in a patient diagnosed with late onset breast and ovarian cancer: implications for genetic testing. Breast Cancer Res Treat 132 (1): 87-95, 2012.
- Diez O, Gutiérrez-Enríquez S, Mediano C, et al.: A novel de novo BRCA2 mutation of paternal origin identified in a Spanish woman with early onset bilateral breast cancer. Breast Cancer Res Treat 121 (1): 221-5, 2010.
- Garcia-Casado Z, Romero I, Fernandez-Serra A, et al.: A de novo complete BRCA1 gene deletion identified in a Spanish woman with early bilateral breast cancer. BMC Med Genet 12: 134, 2011.
- Hansen TV, Bisgaard ML, Jønson L, et al.: Novel de novo BRCA2 mutation in a patient with a family history of breast cancer. BMC Med Genet 9: 58, 2008.
- Kwong A, Ng EK, Tang EY, et al.: A novel de novo BRCA1 mutation in a Chinese woman with early onset breast cancer. Fam Cancer 10 (2): 233-7, 2011.
- Marshall M, Solomon S, Lawrence Wickerham D: Case report: de novo BRCA2 gene mutation in a 35-year-old woman with breast cancer. Clin Genet 76 (5): 427-30, 2009.
- Robson M, Scheuer L, Nafa K, et al.: Unique de novo mutation of BRCA2 in a woman with early onset breast cancer. J Med Genet 39 (2): 126-8, 2002.
- Tesoriero A, Andersen C, Southey M, et al.: De novo BRCA1 mutation in a patient with breast cancer and an inherited BRCA2 mutation. Am J Hum Genet 65 (2): 567-9, 1999.
- van der Luijt RB, van Zon PH, Jansen RP, et al.: De novo recurrent germline mutation of the BRCA2 gene in a patient with early onset breast cancer. J Med Genet 38 (2): 102-5, 2001.
- Morak M, Laner A, Scholz M, et al.: Report on de-novo mutation in the MSH2 gene as a rare event in hereditary nonpolyposis colorectal cancer. Eur J Gastroenterol Hepatol 20 (11): 1101-5, 2008.
- Plasilova M, Zhang J, Okhowat R, et al.: A de novo MLH1 germ line mutation in a 31-year-old colorectal cancer patient. Genes Chromosomes Cancer 45 (12): 1106-10, 2006.
- Win AK, Jenkins MA, Buchanan DD, et al.: Determining the frequency of de novo germline mutations in DNA mismatch repair genes. J Med Genet 48 (8): 530-4, 2011.
- Anderson KG: How well does paternity confidence match actual paternity? Evidence from worldwide nonpaternity rates. Curr Anthropol 47 (3): 513-20, 2006. Also available online. Last accessed November 8, 2023.
- Sasse G, Müller H, Chakraborty R, et al.: Estimating the frequency of nonpaternity in Switzerland. Hum Hered 44 (6): 337-43, 1994 Nov-Dec.
- Voracek M, Haubner T, Fisher ML: Recent decline in nonpaternity rates: a cross-temporal meta-analysis. Psychol Rep 103 (3): 799-811, 2008.
Moderate-Penetrance Genes Associated With Breast and/or Gynecologic Cancers
Background
Pathogenic variants in BRCA1, BRCA2, PALB2, and the genes involved in other rare syndromes discussed in the High-Penetrance Breast and/or Gynecologic Cancer Susceptibility Genes section of this summary account for less than 25% of the familial risk of breast cancer. Despite intensive genetic linkage studies, there do not appear to be other high-penetrance genes that account for a significant fraction of the remaining multiple-case familial clusters. However, several moderate-penetrance genes associated with breast and/or gynecologic cancers have been identified. Genes such as CHEK2 and ATM are associated with a 20% or higher lifetime risk of breast cancer; similarly, genes such as RAD51C, RAD51D, and BRIP1 are associated with a 5% to 10% risk of ovarian cancer. Many of these genes are now included on multigene panels, although the clinical actionability of these findings remains uncertain and under investigation.
Breast and Gynecologic Cancer Susceptibility Genes Identified Through Candidate Gene Approaches
There is a very large literature of genetic epidemiology studies describing associations between various loci and breast cancer risk. Many of these studies suffer from significant design limitations. Perhaps as a consequence, most reported associations do not replicate in follow-up studies. This section is not a comprehensive review of all reported associations. This section describes associations that are believed by the editors to be clinically valid, in that they have been described in several studies or are supported by robust meta-analyses. The clinical utility of these observations remains unclear, however, as the risks associated with these variations usually fall below a threshold that would justify a clinical response.
Fanconi anemia genes
Fanconi anemia (FA) is a rare, inherited condition characterized by bone marrow failure, increased risk of malignancy, and physical abnormalities. To date, 16 FA-related genes, including BRCA1 and BRCA2, have been identified (as outlined in Table 7). FA is mainly an autosomal recessive condition, except when caused by pathogenic variants in FANCB, which is X-linked recessive. FANCA accounts for 60% to 70% of pathogenic variants, FANCC accounts for approximately 14%, and the remaining genes each account for 3% or fewer.
| aRefer to the BRCA1 and BRCA2 summary for information about the cumulative risk of breast cancer in carriers of BRCA1 and BRCA2 pathogenic variants. |
| bRefer to the PALB2 section for information about the cumulative risk of breast cancer in carriers of PALB2 pathogenic variants. |
| cModerate risk is defined as a statistically significant, twofold or lower increased risk estimate. |
| High-Risk Genes |
| – BRCA1 (FANCS)a |
| – BRCA2 (FANCD1)a |
| – PALB2 (FANCN)b |
| Moderate-Risk Genesc |
| – BRIP1 (FANCJ/BACH1) |
| – FANCD2 |
| – RAD51C (FANCO) |
| Genes With Uncertain or No Significantly Increased Risk |
| – FANCA |
| – FANCB |
| – FANCC |
| – FANCE |
| – FANCF |
| – FANCG (XRCC9) |
| – FANCI (KIAA1794) |
| – FANCL |
| – SLX4 (FANCP) |
| – ERCC4 (FANCQ/XPF) |
Progressive bone marrow failure typically occurs in the first decade, with patients often presenting with thrombocytopenia or leucopenia. The incidence of bone marrow failure is 90% by age 40 to 50 years. The incidence is 10% to 30% for hematologic malignancies (primarily acute myeloid leukemia) and 25% to 30% for nonhematologic malignancies (solid tumors, particularly of the head and neck, skin, gastrointestinal [GI] tract, and genital tract). Physical abnormalities, including short stature, abnormal skin pigmentation, radial ray defects (including malformation of the thumbs), abnormalities of the urinary tract, eyes, ears, heart, GI system, and central nervous system, hypogonadism, and developmental delay are present in 60% to 75% of affected individuals.
Variants in some of the FA genes, most notably BRCA1 and BRCA2, but also PALB2, RAD51C, and BRIP1, among others, may predispose to breast cancer in heterozygotes. Given the widespread availability of multigene (panel) tests, genetic testing of many of the FA genes is frequently performed despite uncertain cancer risks and the lack of available evidence-based medical management recommendations for many of these genes.
FA gene pathogenic variant carrier status can have implications for reproductive decision making because pathogenic variants in these genes can lead to serious childhood onset of disease if both parents are carriers of pathogenic variants in the same gene. Partner testing may be considered.
BRIP1
BRIP1 (also known as BACH1) encodes a helicase that interacts with the BRCA1 C-terminal domain. This gene also has a role in BRCA1-dependent DNA repair and cell cycle checkpoint function. Biallelic pathogenic variants in BRIP1 are a cause of FA, much like such pathogenic variants in BRCA2.
Monoallelic pathogenic variants in BRIP1 have emerged as having a significant association with increased ovarian cancer risk. Nine-tenths to two and half percent of women with ovarian cancer carry a pathogenic variant in BRIP1. Odds ratios (ORs) for ovarian cancer in individuals with a BRIP1 pathogenic variant range from 2.2 to 5.0. The median age of ovarian cancer diagnosis in individuals with BRIP1 pathogenic variants ranges from the mid-50s to 70 years. BRIP1 pathogenic variants have been seen in high-grade serous, borderline, and endometrioid ovarian cancers, but not in clear-cell or mucinous types. Per current National Comprehensive Cancer Network (NCCN) guidelines, risk-reducing salpingo-oophorectomy is recommended for women who carry a BRIP1 pathogenic variant.
With respect to breast cancer risk, several studies consistently report ORs less than 2.0. A meta-analysis of 148 studies found an OR for breast cancer of 1.62 in individuals with BRIP1 pathogenic variants (95% confidence interval [CI], 1.20–2.20). ORs for breast cancer in BRIP1 carriers ranged from 0.60 to 1.81 in other studies. There is a growing consensus that BRIP1 is not a moderate- to high-risk breast cancer susceptibility gene. However, studies are looking at the possible associations between BRIP1 pathogenic variants and certain subtypes of breast cancer, such as triple-negative breast cancer. Limitations of these BRIP1 association studies include the following: rarity of BRIP1 pathogenic variants, heterogeneity of study methodologies, and inconsistent reporting of family histories in many of the published studies.
CHEK2
CHEK2 is a gene involved in the DNA damage repair response pathway. Based on numerous studies, a polymorphism, 1100delC, appears to be a rare, moderate-penetrance cancer susceptibility allele. One study identified the pathogenic variant in 1.2% of the European controls, 4.2% of the European BRCA1/BRCA2-negative familial breast cancer cases, and 1.4% of unselected female breast cancer cases. In a group of 1,479 Dutch women younger than 50 years with invasive breast cancer, 3.7% were found to have the CHEK2 1100delC pathogenic variant. In additional European and U.S. (where the pathogenic variant appears to be slightly less common) studies, including a large prospective study, the frequency of CHEK2 pathogenic variants detected in familial breast or ovarian cancer cases has ranged from 0% to 11%; overall, these studies have found an approximately 1.5-fold to 3-fold increased risk of female breast cancer. A multicenter combined analysis and reanalysis of nearly 20,000 subjects from ten case-control studies, however, has verified a significant 2.3-fold excess of breast cancer among carriers of pathogenic variants. A subsequent meta-analysis based on 29,154 cases and 37,064 controls from 25 case-control studies found a significant association between CHEK2 1100delC heterozygotes and breast cancer risk (OR, 2.75; 95% CI, 2.25–3.36). The ORs and CIs in unselected, familial, and early-onset breast cancer subgroups were 2.33 (1.79–3.05), 3.72 (2.61–5.31), and 2.78 (2.28–3.39), respectively. However, study limitations included pooling of populations without subgroup analysis, using a mix of population-based and hospital-based controls, and basing results on unadjusted estimates (as cases and controls were matched on only a few common factors); therefore, results should be interpreted in the context of these limitations. In a series of male breast cancer patients, the CHEK2 1100delC variant was significantly more frequently identified than in controls, suggesting that this variant is also associated with an increased risk of male breast cancer.
Two studies have suggested that the risk associated with a CHEK2 1100delC pathogenic variant was stronger in the families of probands ascertained because of bilateral breast cancer. Furthermore, a meta-analysis of carriers of 1100delC pathogenic variants estimated the risk of breast cancer to be 42% by age 70 years in women with a family history of breast cancer. Similarly, a Polish study reported that CHEK2 truncating pathogenic variants confer breast cancer risks based on a family history of breast cancer as follows: no family history, 20%; one second-degree relative (SDR), 28%; one first-degree relative (FDR), 34%; and both FDRs and SDRs, 44%. Moreover, a Dutch study suggested that female homozygotes for the CHEK2 1100delC variant have a greater-than-twofold increased breast cancer risk compared with heterozygotes. Although there have been conflicting reports regarding cancers other than breast cancer associated with CHEK2 pathogenic variants, this may be dependent on variant type (i.e., missense vs. truncating) or population studied and is not currently of clinical utility. The contribution of CHEK2 variants to breast cancer may depend on the population studied, with a potentially higher variant prevalence in Poland. Carriers of CHEK2 variants in Poland may be more susceptible to estrogen receptor (ER)–positive breast cancer.
A large Dutch study of 86,975 individuals reported an increased risk of cancers other than breast and colon for carriers of the CHEK2 1100delC pathogenic variant, although additional studies are needed to further refine these risks.
(Refer to the CHEK2 section in Genetics of Colorectal Cancer for more information.)
ATM
Ataxia telangiectasia (AT) is an autosomal recessive disorder characterized by neurologic deterioration, telangiectasias, immunodeficiency states, and hypersensitivity to ionizing radiation. It is estimated that 1% of the general population may be heterozygote carriers of ATM variants. More than 300 variants in the gene have been identified, most of which are truncating variants. ATM proteins have been shown to play a role in cell cycle control. In vitro, AT-deficient cells are sensitive to ionizing radiation and radiomimetic drugs, and lack cell cycle regulatory properties after exposure to radiation. There is insufficient evidence to recommend against radiation therapy in carriers of a single ATM pathogenic variant (heterozygotes).
Initial, large epidemiological studies demonstrated a statistically increased relative risk (RR) of approximately 2.0 for breast cancer among female ATM heterozygotes. Subsequent, large international consortium-based studies have refined risk estimates. An international study based on 113,000 females from 25 countries reported an OR of 2.10 (95% CI, 1.71–2.57) for breast cancer in ATM heterozygotes. ATM pathogenic variants were also associated with ER-positive tumors. Domains specifically associated with higher breast cancer risks included the FRAP–ATM–TRRAP (FAT) domain (P = .00019 in all studies) and protein kinase domains (P = .00092 in all studies). Similarly, a United States–based study of 63,000 women reported an OR of 1.82 for breast cancer in ATM heterozygotes (95% CI, 1.46–2.27) and also reported an association between ATM pathogenic variants and ER-positive breast cancers. A similar OR of 2.03 (95% CI, 1.89–2.19) was estimated for invasive ductal breast cancer through a commercial, lab-based study of 4,607 individuals with ATM pathogenic or likely pathogenic variants.
Age-specific cumulative breast cancer risks modeled through a meta-analysis were reported to be 6.02% by age 50 years and 32.83% by age 80 years. Another meta-analysis reported the RR for female breast cancer as 3.0 in ATM carriers (95% CI, 2.1–4.5). A subsequent systematic review and meta-analysis estimated an adjusted OR of 1.67 for breast cancer risk in individuals with ATM pathogenic variants (95% CI, 0.73–3.82) based on seven adjusted case-control studies. The crude OR was 2.27 (95% CI, 1.17–4.40) based on nine unadjusted case-control studies. The RR was estimated as 1.68 (95% CI, 1.17–2.40) based on two cohort studies. Overall, the findings suggested genotype-phenotype correlations, with the ATM c.7271T>G variant (also known as the ATM Val2424Gly variant) as the most predisposing factor and with limited predictive ability for Asp1853Val, Leu546Val, and Ser707Pro ATM variants. Per NCCN guidelines, it is recommended that women who carry an ATM pathogenic variant have annual mammograms starting at age 40 years with consideration of breast magnetic resonance imaging with and without contrast beginning at age 30 to 35 years.
While multiple studies have reported that most ATM pathogenic variants impart moderate risks for breast cancer, the c.7271T>G missense variant has been shown to predispose individuals to higher breast cancer risks. Specifically, in a commercial laboratory, data-based study of patients referred for hereditary cancer testing with a multi-gene panel (N = 627,742) including 4,607 ATM pathogenic or likely pathogenic variant carriers, risk of invasive ductal breast cancer was higher for the c.7271T>G missense variant (OR, 3.76; 95% CI, 2.76–5.12) than for other missense and truncating ATM variants.
Some studies reported an association between ATM and ovarian cancer, with ovarian cancer lifetime risk approaching ~3%. A commercial laboratory, data-based study reported an OR of 1.57 (95% CI, 1.35–1.83) for ovarian cancer in ATM pathogenic variant carriers.
Pancreatic cancer has also been associated with ATM pathogenic variants, with an OR of 4.21 (95% CI, 3.24–5.47) reported through a commercial lab–based study. Among 130 pancreatic cancer kindreds with a germline ATM pathogenic variant, the cumulative risk of pancreatic cancer was 1.1% (95% CI, 0.8%–1.3%) by age 50 years, 6.3% (95% CI, 3.9%–8.7%) by age 70 years, and 9.5% (95% CI, 5.0%–14.0%) by age 80 years. Overall, the RR of pancreatic cancer was 6.5 (95% CI, 4.5–9.5) in ATM pathogenic variant carriers when compared with noncarriers. The average age at diagnosis was 64 years (range, 31–98 y).
The association between ATM pathogenic variants and prostate cancer risk have been inconclusive, with a commercial lab–based study reporting an OR of 2.58 (95% CI, 1.93–3.44). For more information, see the ATM section in Genetics of Prostate Cancer.
RAD51
RAD51 and the family of RAD51-related genes, also known as RAD51 paralogs, are thought to encode proteins that are involved in DNA damage repair through homologous recombination and interaction with numerous other DNA repair proteins, including BRCA1 and BRCA2. The RAD51 protein plays a central role in single-strand annealing in the DNA damage response. RAD51 recruitment to break sites and recombinational DNA repair depend on the RAD51 paralogs, although their precise cellular functions are poorly characterized. Variants in these genes are thought to result in loss of RAD51 focus formation in response to DNA damage.
One of five RAD51-related genes, RAD51C has been reported to be linked to both FA-like disorders and familial breast and ovarian cancers. The literature, however, has produced contradictory findings. In a study of 480 German families characterized by breast and ovarian cancers who were negative for BRCA1 and BRCA2 pathogenic variants, six monoallelic variants in RAD51C were found (frequency of 1.3%). Another study screened 286 BRCA1/BRCA2-negative patients with breast cancer and/or ovarian cancer and found one likely pathogenic variant in RAD51C-G153D. RAD51C pathogenic variants have also been reported in Australian, British, Finnish, and Spanish non-BRCA1/BRCA2 ovarian cancer–only and breast/ovarian cancer families, and in unselected ovarian cancer cases, with frequencies ranging from 0% to 3% in these populations. In a sample of 206 high-risk Jewish women (including 79 of Ashkenazi origin) previously tested for the common Jewish pathogenic variants, two previously described and possibly pathogenic missense variants were detected. Four additional studies were unable to confirm an association between the RAD51C gene and hereditary breast cancer or ovarian cancer.
In addition to carriers of RAD51C pathogenic variants, there are other RAD51 paralogs, including RAD51B, RAD51D, RAD51L1, XRCC2, and XRCC3, that may be associated with breast and/or ovarian cancer risk, although the clinical significance of these findings is unknown. In a case-control study of 3,429 ovarian cancer patients, RAD51C and RAD51D pathogenic variants were more commonly found in ovarian cancer cases (0.82%) than in controls (0.11%, P< .001).
In addition to germline variants, different polymorphisms of RAD51 have been hypothesized to have reduced capacity to repair DNA defects, resulting in increased susceptibility to familial breast cancer. The Consortium of Investigators of Modifiers of BRCA1/BRCA2 (CIMBA) pooled data from 8,512 carriers of BRCA1 and BRCA2 pathogenic variants and found evidence of an increased risk of breast cancer among women who were BRCA2 carriers and who were homozygous for CC at the RAD51 135G→C SNV (hazard ratio, 1.17; 95% CI, 0.91–1.51).
Several meta-analyses have investigated the association between the RAD51 135G→C polymorphism and breast cancer risk. There is significant overlap in the studies reported in these meta-analyses, significant variability in the characteristics of the populations included, and significant methodologic limitations to their findings. A meta-analysis of nine epidemiologic studies involving 13,241 cases and 13,203 controls of unknown BRCA1/BRCA2 status found that women carrying the CC genotype had an increased risk of breast cancer compared with women with the GG or GC genotype (OR, 1.35; 95% CI, 1.04–1.74). A meta-analysis of 14 case-control studies involving 12,183 cases and 10,183 controls confirmed an increased risk only for women who were known BRCA2 carriers (OR, 4.92; 95% CI, 1.10–21.83). Another meta-analysis of 12 studies included only studies of known BRCA-negative cases and found no association between RAD51 135G→C and breast cancer.
In summary, among this conflicting data is substantial evidence for a modest association between germline variants in RAD51C and breast cancer and ovarian cancer. There is also evidence of an association between polymorphisms in RAD51 135G→C among women with homozygous CC genotypes and breast cancer, particularly among BRCA2 carriers. These associations are plausible given the known role of RAD51 in the maintenance of genomic stability.
SMARCA4
SMARCA4 encodes BRG1 and is a catalytic subunit of the SWI/SNF chromatin remodeling complex, which plays a major role in rendering chromatin accessible to regulation of gene expression.
Small cell carcinoma of the ovary, hypercalcemic type (SCCOHT) is a rare, aggressive tumor that has an early age at onset (before age 40 y) and a poor prognosis. Familial clustering is sometimes present. SCCOHT tumors may be unilateral or bilateral and have been characterized histologically by the presence of small hyperchromatic cells with brisk mitotic activity. A multimodality approach including surgery, chemotherapy, and radiation therapy has been suggested for the treatment of SCCOHT. Given the paraneoplastic phenomenon of hypercalcemia in 60% of cases, tracking calcium levels is useful in monitoring the course of disease. With a wide range of differential diagnoses including germ cell tumors, sex cord–stromal tumors, and undifferentiated carcinomas, SCCOHT remains classified by the World Health Organization as a "miscellaneous tumor" but more recently has been sequenced to be a malignant rhabdoid tumor. Through exome sequencing, most cases of SCCOHT have been found to lack functional SMARCA4/BRG1; in fact, pathogenic variants in SMARCA4 may be the sole variants responsible for SCCOHT.
Despite only approximately 300 cases in the literature, three separate research groups showed SCCOHT to be associated with germline pathogenic variants and somatic variants in the SMARCA4 gene. In one study of 12 young women with SCCOHT, sequencing of paired tumor and normal samples identified inactivating biallelic SMARCA4 pathogenic variants in each case. Only four additional nonrecurrent somatic genes were identified in any of the other 278 genes sequenced. Immunohistochemistry demonstrated loss of SMARCA4 protein expression in seven of nine tested cases, consistent with a tumor-suppressor gene function. In a second study of another 12 patients, next-generation sequencing also identified SMARCA4 as the only recurrently variant gene, with the majority of variants predicted to result in a truncated protein. A third study included three families in whom whole-exome sequencing with Sanger sequencing confirmation identified at least one germline pathogenic variant or somatic variant in 24 of 26 cases. Overall, 38 of 43 (88%) of SCCOHT tumors showed loss of SMARCA4 expression, in comparison to only 1 of 139 (0.7%) other ovarian tumor types.
Because of the rarity of this tumor, the penetrance of SMARCA4 is unknown. There is currently no consensus for management, yet SMARCA4 is on the larger multigene panels currently available for genetic testing, and risk-reducing surgery has been offered to pathogenic variant carriers.
References
- Easton DF: How many more breast cancer predisposition genes are there? Breast Cancer Res 1 (1): 14-7, 1999.
- Smith P, McGuffog L, Easton DF, et al.: A genome wide linkage search for breast cancer susceptibility genes. Genes Chromosomes Cancer 45 (7): 646-55, 2006.
- Cybulski C, Wokołorczyk D, Jakubowska A, et al.: Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J Clin Oncol 29 (28): 3747-52, 2011.
- Thompson D, Duedal S, Kirner J, et al.: Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst 97 (11): 813-22, 2005.
- Pelttari LM, Heikkinen T, Thompson D, et al.: RAD51C is a susceptibility gene for ovarian cancer. Hum Mol Genet 20 (16): 3278-88, 2011.
- Loveday C, Turnbull C, Ramsay E, et al.: Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet 43 (9): 879-82, 2011.
- Mehta PA, Tolar J: Fanconi Anemia. In: Adam MP, Feldman J, Mirzaa GM, et al., eds.: GeneReviews. University of Washington, Seattle, 1993-2024, pp. Available online. Last accessed November 8, 2023.
- Levitus M, Waisfisz Q, Godthelp BC, et al.: The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet 37 (9): 934-5, 2005.
- Levran O, Attwooll C, Henry RT, et al.: The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet 37 (9): 931-3, 2005.
- Litman R, Peng M, Jin Z, et al.: BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 8 (3): 255-65, 2005.
- Moyer CL, Ivanovich J, Gillespie JL, et al.: Rare BRIP1 Missense Alleles Confer Risk for Ovarian and Breast Cancer. Cancer Res 80 (4): 857-867, 2020.
- Suszynska M, Ratajska M, Kozlowski P: BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: mutation prevalence and precise risk estimates based on a pooled analysis of ~30,000 cases. J Ovarian Res 13 (1): 50, 2020.
- Lhotova K, Stolarova L, Zemankova P, et al.: Multigene Panel Germline Testing of 1333 Czech Patients with Ovarian Cancer. Cancers (Basel) 12 (4): , 2020.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 2.2024. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed October 31, 2023.
- Alter BP, Best AF: Frequency of heterozygous germline pathogenic variants in genes for Fanconi anemia in patients with non-BRCA1/BRCA2 breast cancer: a meta-analysis. Breast Cancer Res Treat 182 (2): 465-476, 2020.
- Meijers-Heijboer H, van den Ouweland A, Klijn J, et al.: Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet 31 (1): 55-9, 2002.
- Kuschel B, Auranen A, Gregory CS, et al.: Common polymorphisms in checkpoint kinase 2 are not associated with breast cancer risk. Cancer Epidemiol Biomarkers Prev 12 (8): 809-12, 2003.
- Sodha N, Bullock S, Taylor R, et al.: CHEK2 variants in susceptibility to breast cancer and evidence of retention of the wild type allele in tumours. Br J Cancer 87 (12): 1445-8, 2002.
- Ingvarsson S, Sigbjornsdottir BI, Huiping C, et al.: Mutation analysis of the CHK2 gene in breast carcinoma and other cancers. Breast Cancer Res 4 (3): R4, 2002.
- Vahteristo P, Bartkova J, Eerola H, et al.: A CHEK2 genetic variant contributing to a substantial fraction of familial breast cancer. Am J Hum Genet 71 (2): 432-8, 2002.
- Meijers-Heijboer H, Wijnen J, Vasen H, et al.: The CHEK2 1100delC mutation identifies families with a hereditary breast and colorectal cancer phenotype. Am J Hum Genet 72 (5): 1308-14, 2003.
- Schmidt MK, Tollenaar RA, de Kemp SR, et al.: Breast cancer survival and tumor characteristics in premenopausal women carrying the CHEK2*1100delC germline mutation. J Clin Oncol 25 (1): 64-9, 2007.
- Weischer M, Bojesen SE, Tybjaerg-Hansen A, et al.: Increased risk of breast cancer associated with CHEK2*1100delC. J Clin Oncol 25 (1): 57-63, 2007.
- Iniesta MD, Gorin MA, Chien LC, et al.: Absence of CHEK2*1100delC mutation in families with hereditary breast cancer in North America. Cancer Genet Cytogenet 202 (2): 136-40, 2010.
- Offit K, Pierce H, Kirchhoff T, et al.: Frequency of CHEK2*1100delC in New York breast cancer cases and controls. BMC Med Genet 4 (1): 1, 2003.
- Oldenburg RA, Kroeze-Jansema K, Kraan J, et al.: The CHEK2*1100delC variant acts as a breast cancer risk modifier in non-BRCA1/BRCA2 multiple-case families. Cancer Res 63 (23): 8153-7, 2003.
- Neuhausen S, Dunning A, Steele L, et al.: Role of CHEK2*1100delC in unselected series of non-BRCA1/2 male breast cancers. Int J Cancer 108 (3): 477-8, 2004.
- Ohayon T, Gal I, Baruch RG, et al.: CHEK2*1100delC and male breast cancer risk in Israel. Int J Cancer 108 (3): 479-80, 2004.
- CHEK2 Breast Cancer Case-Control Consortium: CHEK2*1100delC and susceptibility to breast cancer: a collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am J Hum Genet 74 (6): 1175-82, 2004.
- Yang Y, Zhang F, Wang Y, et al.: CHEK2 1100delC variant and breast cancer risk in Caucasians: a meta-analysis based on 25 studies with 29,154 cases and 37,064 controls. Asian Pac J Cancer Prev 13 (7): 3501-5, 2012.
- Hallamies S, Pelttari LM, Poikonen-Saksela P, et al.: CHEK2 c.1100delC mutation is associated with an increased risk for male breast cancer in Finnish patient population. BMC Cancer 17 (1): 620, 2017.
- Johnson N, Fletcher O, Naceur-Lombardelli C, et al.: Interaction between CHEK2*1100delC and other low-penetrance breast-cancer susceptibility genes: a familial study. Lancet 366 (9496): 1554-7, 2005 Oct 29-Nov 4.
- Fletcher O, Johnson N, Dos Santos Silva I, et al.: Family history, genetic testing, and clinical risk prediction: pooled analysis of CHEK2 1100delC in 1,828 bilateral breast cancers and 7,030 controls. Cancer Epidemiol Biomarkers Prev 18 (1): 230-4, 2009.
- Weischer M, Bojesen SE, Ellervik C, et al.: CHEK2*1100delC genotyping for clinical assessment of breast cancer risk: meta-analyses of 26,000 patient cases and 27,000 controls. J Clin Oncol 26 (4): 542-8, 2008.
- Adank MA, Jonker MA, Kluijt I, et al.: CHEK2*1100delC homozygosity is associated with a high breast cancer risk in women. J Med Genet 48 (12): 860-3, 2011.
- Gronwald J, Cybulski C, Piesiak W, et al.: Cancer risks in first-degree relatives of CHEK2 mutation carriers: effects of mutation type and cancer site in proband. Br J Cancer 100 (9): 1508-12, 2009.
- Wasielewski M, den Bakker MA, van den Ouweland A, et al.: CHEK2 1100delC and male breast cancer in the Netherlands. Breast Cancer Res Treat 116 (2): 397-400, 2009.
- Osorio A, Rodríguez-López R, Díez O, et al.: The breast cancer low-penetrance allele 1100delC in the CHEK2 gene is not present in Spanish familial breast cancer population. Int J Cancer 108 (1): 54-6, 2004.
- Syrjäkoski K, Kuukasjärvi T, Auvinen A, et al.: CHEK2 1100delC is not a risk factor for male breast cancer population. Int J Cancer 108 (3): 475-6, 2004.
- Tsou HC, Teng DH, Ping XL, et al.: The role of MMAC1 mutations in early-onset breast cancer: causative in association with Cowden syndrome and excluded in BRCA1-negative cases. Am J Hum Genet 61 (5): 1036-43, 1997.
- Olopade OI, Weber BL: Breast cancer genetics: toward molecular characterization of individuals at increased risk for breast cancer: part I. Cancer: Principles and Practice of Oncology Updates 12 (10): 1-12, 1998.
- Cybulski C, Górski B, Huzarski T, et al.: CHEK2-positive breast cancers in young Polish women. Clin Cancer Res 12 (16): 4832-5, 2006.
- Cybulski C, Huzarski T, Byrski T, et al.: Estrogen receptor status in CHEK2-positive breast cancers: implications for chemoprevention. Clin Genet 75 (1): 72-8, 2009.
- Näslund-Koch C, Nordestgaard BG, Bojesen SE: Increased Risk for Other Cancers in Addition to Breast Cancer for CHEK2*1100delC Heterozygotes Estimated From the Copenhagen General Population Study. J Clin Oncol 34 (11): 1208-16, 2016.
- Savitsky K, Bar-Shira A, Gilad S, et al.: A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268 (5218): 1749-53, 1995.
- Telatar M, Teraoka S, Wang Z, et al.: Ataxia-telangiectasia: identification and detection of founder-effect mutations in the ATM gene in ethnic populations. Am J Hum Genet 62 (1): 86-97, 1998.
- Uhrhammer N, Bay JO, Bignon YJ: Seventh International Workshop on Ataxia-Telangiectasia. Cancer Res 58 (15): 3480-5, 1998.
- Ahmed M, Rahman N: ATM and breast cancer susceptibility. Oncogene 25 (43): 5906-11, 2006.
- Khanna KK, Chenevix-Trench G: ATM and genome maintenance: defining its role in breast cancer susceptibility. J Mammary Gland Biol Neoplasia 9 (3): 247-62, 2004.
- Gilad S, Chessa L, Khosravi R, et al.: Genotype-phenotype relationships in ataxia-telangiectasia and variants. Am J Hum Genet 62 (3): 551-61, 1998.
- Renwick A, Thompson D, Seal S, et al.: ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet 38 (8): 873-5, 2006.
- Dorling L, Carvalho S, Allen J, et al.: Breast Cancer Risk Genes - Association Analysis in More than 113,000 Women. N Engl J Med 384 (5): 428-439, 2021.
- Hu C, Hart SN, Gnanaolivu R, et al.: A Population-Based Study of Genes Previously Implicated in Breast Cancer. N Engl J Med 384 (5): 440-451, 2021.
- Hall MJ, Bernhisel R, Hughes E, et al.: Germline Pathogenic Variants in the Ataxia Telangiectasia Mutated (ATM) Gene are Associated with High and Moderate Risks for Multiple Cancers. Cancer Prev Res (Phila) 14 (4): 433-440, 2021.
- Marabelli M, Cheng SC, Parmigiani G: Penetrance of ATM Gene Mutations in Breast Cancer: A Meta-Analysis of Different Measures of Risk. Genet Epidemiol 40 (5): 425-31, 2016.
- van Os NJ, Roeleveld N, Weemaes CM, et al.: Health risks for ataxia-telangiectasia mutated heterozygotes: a systematic review, meta-analysis and evidence-based guideline. Clin Genet 90 (2): 105-17, 2016.
- Moslemi M, Moradi Y, Dehghanbanadaki H, et al.: The association between ATM variants and risk of breast cancer: a systematic review and meta-analysis. BMC Cancer 21 (1): 27, 2021.
- Bernstein JL, Teraoka S, Southey MC, et al.: Population-based estimates of breast cancer risks associated with ATM gene variants c.7271T>G and c.1066-6T>G (IVS10-6T>G) from the Breast Cancer Family Registry. Hum Mutat 27 (11): 1122-8, 2006.
- Goldgar DE, Healey S, Dowty JG, et al.: Rare variants in the ATM gene and risk of breast cancer. Breast Cancer Res 13 (4): R73, 2011.
- Lu HM, Li S, Black MH, et al.: Association of Breast and Ovarian Cancers With Predisposition Genes Identified by Large-Scale Sequencing. JAMA Oncol 5 (1): 51-57, 2019.
- Pennington KP, Walsh T, Harrell MI, et al.: Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res 20 (3): 764-75, 2014.
- Hsu FC, Roberts NJ, Childs E, et al.: Risk of Pancreatic Cancer Among Individuals With Pathogenic Variants in the ATM Gene. JAMA Oncol 7 (11): 1664-1668, 2021.
- Suwaki N, Klare K, Tarsounas M: RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin Cell Dev Biol 22 (8): 898-905, 2011.
- Vaz F, Hanenberg H, Schuster B, et al.: Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet 42 (5): 406-9, 2010.
- Meindl A, Hellebrand H, Wiek C, et al.: Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet 42 (5): 410-4, 2010.
- Clague J, Wilhoite G, Adamson A, et al.: RAD51C germline mutations in breast and ovarian cancer cases from high-risk families. PLoS One 6 (9): e25632, 2011.
- Thompson ER, Boyle SE, Johnson J, et al.: Analysis of RAD51C germline mutations in high-risk breast and ovarian cancer families and ovarian cancer patients. Hum Mutat 33 (1): 95-9, 2012.
- Vuorela M, Pylkäs K, Hartikainen JM, et al.: Further evidence for the contribution of the RAD51C gene in hereditary breast and ovarian cancer susceptibility. Breast Cancer Res Treat 130 (3): 1003-10, 2011.
- Romero A, Pérez-Segura P, Tosar A, et al.: A HRM-based screening method detects RAD51C germ-line deleterious mutations in Spanish breast and ovarian cancer families. Breast Cancer Res Treat 129 (3): 939-46, 2011.
- Osorio A, Endt D, Fernández F, et al.: Predominance of pathogenic missense variants in the RAD51C gene occurring in breast and ovarian cancer families. Hum Mol Genet 21 (13): 2889-98, 2012.
- Ramus SJ, Song H, Dicks E, et al.: Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J Natl Cancer Inst 107 (11): , 2015.
- Blanco A, Gutiérrez-Enríquez S, Santamariña M, et al.: RAD51C germline mutations found in Spanish site-specific breast cancer and breast-ovarian cancer families. Breast Cancer Res Treat 147 (1): 133-43, 2014.
- Norquist BM, Harrell MI, Brady MF, et al.: Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol 2 (4): 482-90, 2016.
- Kushnir A, Laitman Y, Shimon SP, et al.: Germline mutations in RAD51C in Jewish high cancer risk families. Breast Cancer Res Treat 136 (3): 869-74, 2012.
- Wong MW, Nordfors C, Mossman D, et al.: BRIP1, PALB2, and RAD51C mutation analysis reveals their relative importance as genetic susceptibility factors for breast cancer. Breast Cancer Res Treat 127 (3): 853-9, 2011.
- Zheng Y, Zhang J, Hope K, et al.: Screening RAD51C nucleotide alterations in patients with a family history of breast and ovarian cancer. Breast Cancer Res Treat 124 (3): 857-61, 2010.
- Akbari MR, Tonin P, Foulkes WD, et al.: RAD51C germline mutations in breast and ovarian cancer patients. Breast Cancer Res 12 (4): 404, 2010.
- De Leeneer K, Van Bockstal M, De Brouwer S, et al.: Evaluation of RAD51C as cancer susceptibility gene in a large breast-ovarian cancer patient population referred for genetic testing. Breast Cancer Res Treat 133 (1): 393-8, 2012.
- Thomas G, Jacobs KB, Kraft P, et al.: A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1). Nat Genet 41 (5): 579-84, 2009.
- Figueroa JD, Garcia-Closas M, Humphreys M, et al.: Associations of common variants at 1p11.2 and 14q24.1 (RAD51L1) with breast cancer risk and heterogeneity by tumor subtype: findings from the Breast Cancer Association Consortium. Hum Mol Genet 20 (23): 4693-706, 2011.
- Osher DJ, De Leeneer K, Michils G, et al.: Mutation analysis of RAD51D in non-BRCA1/2 ovarian and breast cancer families. Br J Cancer 106 (8): 1460-3, 2012.
- Pelttari LM, Kiiski J, Nurminen R, et al.: A Finnish founder mutation in RAD51D: analysis in breast, ovarian, prostate, and colorectal cancer. J Med Genet 49 (7): 429-32, 2012.
- Golmard L, Castéra L, Krieger S, et al.: Contribution of germline deleterious variants in the RAD51 paralogs to breast and ovarian cancers. Eur J Hum Genet 25 (12): 1345-1353, 2017.
- Song H, Dicks E, Ramus SJ, et al.: Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J Clin Oncol 33 (26): 2901-7, 2015.
- Antoniou AC, Sinilnikova OM, Simard J, et al.: RAD51 135G-->C modifies breast cancer risk among BRCA2 mutation carriers: results from a combined analysis of 19 studies. Am J Hum Genet 81 (6): 1186-200, 2007.
- He XF, Su J, Zhang Y, et al.: Need for clarification of data in the recent meta-analysis about RAD51 135G>C polymorphism and breast cancer risk. Breast Cancer Res Treat 129 (2): 649-51; author reply 652-3, 2011.
- Lu W, Wang X, Lin H, et al.: Mutation screening of RAD51C in high-risk breast and ovarian cancer families. Fam Cancer 11 (3): 381-5, 2012.
- Wang WW, Spurdle AB, Kolachana P, et al.: A single nucleotide polymorphism in the 5' untranslated region of RAD51 and risk of cancer among BRCA1/2 mutation carriers. Cancer Epidemiol Biomarkers Prev 10 (9): 955-60, 2001.
- Wang Z, Dong H, Fu Y, et al.: RAD51 135G>C polymorphism contributes to breast cancer susceptibility: a meta-analysis involving 26,444 subjects. Breast Cancer Res Treat 124 (3): 765-9, 2010.
- Zhou GW, Hu J, Peng XD, et al.: RAD51 135G>C polymorphism and breast cancer risk: a meta-analysis. Breast Cancer Res Treat 125 (2): 529-35, 2011.
- Yu KD, Yang C, Fan L, et al.: RAD51 135G>C does not modify breast cancer risk in non-BRCA1/2 mutation carriers: evidence from a meta-analysis of 12 studies. Breast Cancer Res Treat 126 (2): 365-71, 2011.
- Dickersin GR, Kline IW, Scully RE: Small cell carcinoma of the ovary with hypercalcemia: a report of eleven cases. Cancer 49 (1): 188-97, 1982.
- Harrison ML, Hoskins P, du Bois A, et al.: Small cell of the ovary, hypercalcemic type -- analysis of combined experience and recommendation for management. A GCIG study. Gynecol Oncol 100 (2): 233-8, 2006.
- Callegaro-Filho D, Gershenson DM, Nick AM, et al.: Small cell carcinoma of the ovary-hypercalcemic type (SCCOHT): A review of 47 cases. Gynecol Oncol 140 (1): 53-7, 2016.
- Foulkes WD, Clarke BA, Hasselblatt M, et al.: No small surprise - small cell carcinoma of the ovary, hypercalcaemic type, is a malignant rhabdoid tumour. J Pathol 233 (3): 209-14, 2014.
- Jelinic P, Mueller JJ, Olvera N, et al.: Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet 46 (5): 424-6, 2014.
- Ramos P, Karnezis AN, Craig DW, et al.: Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet 46 (5): 427-9, 2014.
- Witkowski L, Carrot-Zhang J, Albrecht S, et al.: Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet 46 (5): 438-43, 2014.
- Berchuck A, Witkowski L, Hasselblatt M, et al.: Prophylactic oophorectomy for hereditary small cell carcinoma of the ovary, hypercalcemic type. Gynecol Oncol Rep 12: 20-2, 2015.
Single Nucleotide Variant–Associated Cancer Risks
Polymorphisms underlying polygenic susceptibility to breast and gynecologic cancers are considered low penetrance, a term often applied to sequence variants associated with a minimal to moderate risk. This is in contrast to high-penetrance variants or alleles that are typically associated with more severe phenotypes, for example BRCA1/BRCA2 pathogenic variants leading to an autosomal dominant inheritance pattern in a family, and moderate-penetrance variants such as BRIP1, CHEK2, and RAD51C. (Refer to the High-Penetrance Breast and/or Gynecologic Cancer Susceptibility Genes and the Moderate-Penetrance Genes Associated With Breast and/or Gynecologic Cancer sections of this summary for more information.) Because these types of sequence variants (also called low-penetrance genes, alleles, variants, and polymorphisms) are relatively common in the general population, their overall contribution to cancer risk is estimated to be much greater than the attributable risk in the population from pathogenic variants in BRCA1 and BRCA2. For example, it is estimated by segregation analysis that half of all breast cancer occurs in 12% of the population that is deemed most susceptible. There are no known low-penetrance variants in BRCA1/BRCA2. The N372H variation in BRCA2, initially thought to be a low-penetrance allele, was not verified in a large combined analysis.
Two strategies have attempted to identify low-penetrance polymorphisms leading to breast cancer susceptibility: candidate gene and genome-wide searches. Both involve the epidemiologic case-control study design. The candidate gene approach involves selecting genes based on their known or presumed biological function, relevance to carcinogenesis or organ physiology, and then searching for or testing known genetic variants for an association with cancer risk. This strategy relies on imperfect and incomplete biological knowledge, and, despite some confirmed associations (described below), has been relatively disappointing. The candidate gene approach has largely been replaced by genome-wide association studies (GWAS) in which a very large number of single nucleotide variants(SNVs) (approximately 1 million to 5 million) are chosen within the genome and tested, mostly without regard to their possible biological function, but instead to more uniformly capture all genetic variation throughout the genome.
Genome-Wide Searches
In contrast to assessing candidate genes and/or alleles, GWAS involve comparing a very large set of genetic variants spread throughout the genome. The current paradigm uses sets of as many as 5 million SNVs that are chosen to capture a large portion of common variation within the genome based on the HapMap and the 1000 Genomes Project. By comparing allele frequencies between a large number of cases and controls, typically 1,000 or more of each, and validating promising signals in replication sets of subjects, very robust statistical signals of association have been obtained. The strong correlation between many SNVs that are physically close to each other on the chromosome (linkage disequilibrium) allows one to “scan” the genome for susceptibility alleles even if the biologically relevant variant is not within the tested set of SNVs. Although this between-SNV correlation allows one to interrogate the majority of the genome without having to assay every SNV, when a validated association is obtained, it is not usually obvious which of the many correlated variants is causal.
Genome-wide searches are showing great promise in identifying common, low-penetrance susceptibility alleles for many complex diseases, including breast cancer. The first study involved an initial scan in familial breast cancer cases followed by replication in two large sample sets of sporadic breast cancer, the final being a collection of over 20,000 cases and 20,000 controls from the Breast Cancer Association Consortium (BCAC). Five distinct genomic regions were identified that were within or near the FGFR2, TNRC9, MAP3K1, and LSP1 genes or at the chromosome 8q region. The 8q region and others may harbor multiple independent loci associated with risk. Subsequent genome-wide studies have replicated these loci and identified additional ones. Numerous SNVs identified through large studies of sporadic breast cancer appear to be associated more strongly with estrogen receptor (ER)–positive disease; however, some are associated primarily or exclusively with other subtypes, including triple-negative disease. An online catalog is available of SNV-trait associations from published GWAS for use in investigating genomic characteristics of trait/disease-associated SNVs.
Although the statistical evidence for an association between genetic variation at these loci and breast and ovarian cancer risk is overwhelming, the biologically relevant variants and the mechanism by which they lead to increased risk are unknown and will require further genetic and functional characterization. Additionally, these loci are associated with very modest risk (typically, an odds ratio [OR] <1.5), with more risk variants likely to be identified. No interaction between the SNVs and epidemiologic risk factors for breast cancer have been identified. Furthermore, theoretical models have suggested that common moderate-risk SNVs have limited potential to improve models for individualized risk assessment. These models used receiver operating characteristic (ROC) curve analysis to calculate the area under the curve (AUC) as a measure of discriminatory accuracy. A subsequent study used ROC curve analysis to examine the utility of SNVs in a clinical dataset of more than 5,500 breast cancer cases and nearly 6,000 controls, using a model with traditional risk factors compared with a model using both standard risk factors and ten previously identified SNVs. The addition of genetic information modestly changed the AUC from 58% to 61.8%, a result that was not felt to be clinically significant. Despite this, 32.5% of patients were in a higher quintile of breast cancer risk when genetic information was included, and 20.4% were in a lower quintile of risk. Whether such information has clinical utility is unclear.
More limited data are available regarding ovarian cancer risk. Three GWAS involving staged analysis of more than 10,000 cases and 13,000 controls have been carried out for ovarian cancer. As in other GWAS, the ORs are modest, generally about 1.2 or weaker but implicate a number of genes with plausible biological ties to ovarian cancer, such as BABAM1, whose protein complexes with and may regulate BRCA1, and TIRAPR, which codes for a poly (ADP-ribose) polymerase, molecules that may be important in BRCA1/BRCA2-deficient cells.
Polygenic risk scores for breast and ovarian cancer
The collective influence of many genetic variants has more recently been evaluated using an aggregate score. In 2015, a polygenic risk score (PRS) comprising all of the known breast cancer risk genetic variants or SNVs was estimated in women of European ancestry using 41 studies in the BCAC, including more than 33,000 breast cancer cases and 33,000 controls. This early attempt at estimating a PRS for breast cancer included 77 SNVs, which collectively conferred lifetime risks of developing breast cancer by age 80 years of 3.5% and 29% for women in the lowest and highest 1% of the PRS, respectively. Since then, PRSs incorporating additional genetic variants and examining other breast cancer–related outcomes including tumor and pathological characteristics, mode of detection, and contralateral breast cancer (CBC) have been estimated. In 2019, the PRS with the highest discriminatory ability to date was developed and prospectively validated in the largest GWAS datasets available (79 studies in BCAC and more than 190,000 women in the U.K. Biobank), which incorporates information on 313 genetic variants and is optimized for ER-positive and ER-negative breast cancer. Compared with women in the middle quintile, those in the highest 1% of PRS313 had 4.04-, 4.37-, and 2.78-fold risks of developing breast cancer overall, ER-positive disease, and ER-negative disease, respectively. Lifetime absolute risk (AR) of breast cancer by age 80 years for women in the lowest and highest 1% of PRS313 ranged from 2% to 31% for ER-positive breast cancer, while for ER-negative disease, the ARs ranged from 0.55% to 4%.
Common genomic variants associated with the development of a first primary breast cancer are also associated with the development of CBC. Women in the highest quartile of the PRS had a 1.6-fold increased risk of developing CBC compared with the lowest quartile. Moreover, PRSs of breast and ovarian cancers have been assessed in women who are carriers of BRCA1 and BRCA2 pathogenic variants, and have been found to be predictive of cancer risk in these women, supporting the hypothesis of a shared polygenic component of cancer risk between the general population and variant carriers. The PRS for ER-negative disease had the strongest association with breast cancer risk in BRCA1 variant carriers, while the strongest association in BRCA2 variant carriers was seen for the overall breast cancer PRS. BRCA1 variant carriers had cumulative lifetime risks of 56% and 75% of developing breast cancer at the 10th and 90th percentile of the PRS, respectively. The ovarian cancer PRS was strongly associated with risk for both BRCA1 and BRCA2 variant carriers. For BRCA2 variant carriers, the ovarian cancer risk was 6% and 19% by age 80 years for those at the 10th and 90th percentile of PRS, respectively. The authors noted that the incorporation of the PRS into risk prediction models may better inform decisions on cancer risk management for this population.
Two large studies have supported that PRSs can improve breast cancer risk stratification. PRSs were most important in the breast cancer risk stratification of individuals with CHEK2 and ATM pathogenic variants. After PRSs were incorporated, 30% of individuals with a CHEK2 pathogenic variant and nearly half of the individuals with an ATM pathogenic variant dipped below a 20% lifetime risk of breast cancer. This is significant, since lifetime risk values greater than 20% can prompt more frequent breast cancer screening and other types of clinical management. PRSs were also effective when stratifying breast cancer risk in noncarriers. Gallagher et al. analyzed case-control data from 150,962 women who had multigene hereditary cancer genetic testing. This study examined the impact of a PRS with 86 SNVs on individuals with pathogenic variants in BRCA1, BRCA2, CHEK2, ATM, and PALB2. The PRS was predictive of breast cancer in individuals with pathogenic variants. However, breast cancer risk stratification was more pronounced in noncarriers (OR, 1.47; 95% confidence interval [CI], 1.45–1.49) and CHEK2 pathogenic variant carriers (OR, 1.49; 95% CI, 1.36–1.64) than in carriers of BRCA1 (OR, 1.20; 95% CI, 1.10–1.32) or BRCA2 (OR, 1.23; 95% CI, 1.12–1.34) pathogenic variants. The ORs for ATM (OR, 1.37; 95% CI, 1.21–1.55) and PALB2 (OR, 1.34; 95% CI, 1.16–1.55) pathogenic variant carriers were intermediate when compared with those of BRCA1/2 pathogenic variant carriers, CHEK2 pathogenic variant carriers, and noncarriers. Even though the PRS improved breast cancer risk stratification across all groups, the PRS was most important for individuals with CHEK2 pathogenic variants and for noncarriers. Similarly, Gao et al. analyzed case-control data from 26,798 non-Hispanic White individuals with breast cancer and 26,127 controls using a PRS based on 105 SNVs. More than 95% of BRCA1, BRCA2, and PALB2 pathogenic variant carriers had a 20% lifetime risk of breast cancer. In contrast, 52.5% of ATM pathogenic variant carriers and 69.7% of CHEK2 pathogenic variant carriers without first-degree relatives (FDRs) with breast cancer had a 20% lifetime risk of breast cancer. This was also true in 78.8% of ATM carriers and 89.9% of CHEK2 carriers with an FDR with breast cancer.
Several studies have also examined the extent to which clinical breast cancer risk prediction models can be improved by including information on known susceptibility SNVs, and reporting improved discriminatory accuracy after inclusion of the PRS. For example, in a study combining PRS77 with clinical models, the AUC for predicting breast cancer before age 50 years improved by more than 20%. Clinical trials, including WISDOM and MyPeBs, are in progress to study the potential clinical utility of the PRS for making screening decisions and understanding outcomes. Because PRSs have been largely developed and validated in populations of European ancestry, the utility and prediction accuracy of these PRSs in non-European populations is unknown.
A large study examined whether known reproductive and lifestyle risk factors interact with PRSs to increase breast cancer risk and did not find a multiplicative interaction with established risk factors.
Whole-Genome and Whole-Exome Sequencing
In addition to GWAS interrogating common genetic variants, sequencing-based studies involving whole-genome or whole-exome sequencing are also identifying genes associated with breast cancer, such as XRCC2, a rare, moderate-penetrance breast cancer susceptibility gene. (Refer to the Clinical Sequencing section in Cancer Genetics Overview for more information about whole-exome sequencing.)
References
- Pharoah PD, Antoniou A, Bobrow M, et al.: Polygenic susceptibility to breast cancer and implications for prevention. Nat Genet 31 (1): 33-6, 2002.
- Breast Cancer Association Consortium: Commonly studied single-nucleotide polymorphisms and breast cancer: results from the Breast Cancer Association Consortium. J Natl Cancer Inst 98 (19): 1382-96, 2006.
- Dunning AM, Healey CS, Pharoah PD, et al.: A systematic review of genetic polymorphisms and breast cancer risk. Cancer Epidemiol Biomarkers Prev 8 (10): 843-54, 1999.
- Thorisson GA, Smith AV, Krishnan L, et al.: The International HapMap Project Web site. Genome Res 15 (11): 1592-3, 2005.
- Clarke L, Zheng-Bradley X, Smith R, et al.: The 1000 Genomes Project: data management and community access. Nat Methods 9 (5): 459-62, 2012.
- Evans DM, Cardon LR: Genome-wide association: a promising start to a long race. Trends Genet 22 (7): 350-4, 2006.
- Cardon LR: Genetics. Delivering new disease genes. Science 314 (5804): 1403-5, 2006.
- Chanock SJ, Manolio T, Boehnke M, et al.: Replicating genotype-phenotype associations. Nature 447 (7145): 655-60, 2007.
- Wellcome Trust Case Control Consortium: Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447 (7145): 661-78, 2007.
- Easton DF, Pooley KA, Dunning AM, et al.: Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447 (7148): 1087-93, 2007.
- Stacey SN, Manolescu A, Sulem P, et al.: Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet 39 (7): 865-9, 2007.
- Hunter DJ, Kraft P, Jacobs KB, et al.: A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet 39 (7): 870-4, 2007.
- Turnbull C, Ahmed S, Morrison J, et al.: Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet 42 (6): 504-7, 2010.
- Gold B, Kirchhoff T, Stefanov S, et al.: Genome-wide association study provides evidence for a breast cancer risk locus at 6q22.33. Proc Natl Acad Sci U S A 105 (11): 4340-5, 2008.
- Zheng W, Long J, Gao YT, et al.: Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat Genet 41 (3): 324-8, 2009.
- Kibriya MG, Jasmine F, Argos M, et al.: A pilot genome-wide association study of early-onset breast cancer. Breast Cancer Res Treat 114 (3): 463-77, 2009.
- Murabito JM, Rosenberg CL, Finger D, et al.: A genome-wide association study of breast and prostate cancer in the NHLBI's Framingham Heart Study. BMC Med Genet 8 (Suppl 1): S6, 2007.
- Stacey SN, Manolescu A, Sulem P, et al.: Common variants on chromosome 5p12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet 40 (6): 703-6, 2008.
- Ahmed S, Thomas G, Ghoussaini M, et al.: Newly discovered breast cancer susceptibility loci on 3p24 and 17q23.2. Nat Genet 41 (5): 585-90, 2009.
- Reeves GK, Travis RC, Green J, et al.: Incidence of breast cancer and its subtypes in relation to individual and multiple low-penetrance genetic susceptibility loci. JAMA 304 (4): 426-34, 2010.
- Haiman CA, Chen GK, Vachon CM, et al.: A common variant at the TERT-CLPTM1L locus is associated with estrogen receptor-negative breast cancer. Nat Genet 43 (12): 1210-4, 2011.
- Stevens KN, Fredericksen Z, Vachon CM, et al.: 19p13.1 is a triple-negative-specific breast cancer susceptibility locus. Cancer Res 72 (7): 1795-803, 2012.
- Campa D, Kaaks R, Le Marchand L, et al.: Interactions between genetic variants and breast cancer risk factors in the breast and prostate cancer cohort consortium. J Natl Cancer Inst 103 (16): 1252-63, 2011.
- Milne RL, Gaudet MM, Spurdle AB, et al.: Assessing interactions between the associations of common genetic susceptibility variants, reproductive history and body mass index with breast cancer risk in the breast cancer association consortium: a combined case-control study. Breast Cancer Res 12 (6): R110, 2010.
- Pharoah PD, Antoniou AC, Easton DF, et al.: Polygenes, risk prediction, and targeted prevention of breast cancer. N Engl J Med 358 (26): 2796-803, 2008.
- Gail MH: Discriminatory accuracy from single-nucleotide polymorphisms in models to predict breast cancer risk. J Natl Cancer Inst 100 (14): 1037-41, 2008.
- Gail MH: Value of adding single-nucleotide polymorphism genotypes to a breast cancer risk model. J Natl Cancer Inst 101 (13): 959-63, 2009.
- Wacholder S, Hartge P, Prentice R, et al.: Performance of common genetic variants in breast-cancer risk models. N Engl J Med 362 (11): 986-93, 2010.
- Song H, Ramus SJ, Tyrer J, et al.: A genome-wide association study identifies a new ovarian cancer susceptibility locus on 9p22.2. Nat Genet 41 (9): 996-1000, 2009.
- Goode EL, Chenevix-Trench G, Song H, et al.: A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat Genet 42 (10): 874-9, 2010.
- Bolton KL, Tyrer J, Song H, et al.: Common variants at 19p13 are associated with susceptibility to ovarian cancer. Nat Genet 42 (10): 880-4, 2010.
- Mavaddat N, Pharoah PD, Michailidou K, et al.: Prediction of breast cancer risk based on profiling with common genetic variants. J Natl Cancer Inst 107 (5): , 2015.
- Curtit E, Pivot X, Henriques J, et al.: Assessment of the prognostic role of a 94-single nucleotide polymorphisms risk score in early breast cancer in the SIGNAL/PHARE prospective cohort: no correlation with clinico-pathological characteristics and outcomes. Breast Cancer Res 19 (1): 98, 2017.
- Cuzick J, Brentnall AR, Segal C, et al.: Impact of a Panel of 88 Single Nucleotide Polymorphisms on the Risk of Breast Cancer in High-Risk Women: Results From Two Randomized Tamoxifen Prevention Trials. J Clin Oncol 35 (7): 743-750, 2017.
- Khera AV, Chaffin M, Aragam KG, et al.: Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 50 (9): 1219-1224, 2018.
- Kuchenbaecker KB, McGuffog L, Barrowdale D, et al.: Evaluation of Polygenic Risk Scores for Breast and Ovarian Cancer Risk Prediction in BRCA1 and BRCA2 Mutation Carriers. J Natl Cancer Inst 109 (7): , 2017.
- Li H, Feng B, Miron A, et al.: Breast cancer risk prediction using a polygenic risk score in the familial setting: a prospective study from the Breast Cancer Family Registry and kConFab. Genet Med 19 (1): 30-35, 2017.
- Li J, Ugalde-Morales E, Wen WX, et al.: Differential Burden of Rare and Common Variants on Tumor Characteristics, Survival, and Mode of Detection in Breast Cancer. Cancer Res 78 (21): 6329-6338, 2018.
- Mavaddat N, Michailidou K, Dennis J, et al.: Polygenic Risk Scores for Prediction of Breast Cancer and Breast Cancer Subtypes. Am J Hum Genet 104 (1): 21-34, 2019.
- Robson ME, Reiner AS, Brooks JD, et al.: Association of Common Genetic Variants With Contralateral Breast Cancer Risk in the WECARE Study. J Natl Cancer Inst 109 (10): , 2017.
- Gao C, Polley EC, Hart SN, et al.: Risk of Breast Cancer Among Carriers of Pathogenic Variants in Breast Cancer Predisposition Genes Varies by Polygenic Risk Score. J Clin Oncol 39 (23): 2564-2573, 2021.
- Gallagher S, Hughes E, Wagner S, et al.: Association of a Polygenic Risk Score With Breast Cancer Among Women Carriers of High- and Moderate-Risk Breast Cancer Genes. JAMA Netw Open 3 (7): e208501, 2020.
- Allman R, Dite GS, Hopper JL, et al.: SNPs and breast cancer risk prediction for African American and Hispanic women. Breast Cancer Res Treat 154 (3): 583-9, 2015.
- Dite GS, MacInnis RJ, Bickerstaffe A, et al.: Breast Cancer Risk Prediction Using Clinical Models and 77 Independent Risk-Associated SNPs for Women Aged Under 50 Years: Australian Breast Cancer Family Registry. Cancer Epidemiol Biomarkers Prev 25 (2): 359-65, 2016.
- Shieh Y, Hu D, Ma L, et al.: Breast cancer risk prediction using a clinical risk model and polygenic risk score. Breast Cancer Res Treat 159 (3): 513-25, 2016.
- Starlard-Davenport A, Allman R, Dite GS, et al.: Validation of a genetic risk score for Arkansas women of color. PLoS One 13 (10): e0204834, 2018.
- van Veen EM, Brentnall AR, Byers H, et al.: Use of Single-Nucleotide Polymorphisms and Mammographic Density Plus Classic Risk Factors for Breast Cancer Risk Prediction. JAMA Oncol 4 (4): 476-482, 2018.
- Zhang X, Rice M, Tworoger SS, et al.: Addition of a polygenic risk score, mammographic density, and endogenous hormones to existing breast cancer risk prediction models: A nested case-control study. PLoS Med 15 (9): e1002644, 2018.
- Esserman LJ; WISDOM Study and Athena Investigators: The WISDOM Study: breaking the deadlock in the breast cancer screening debate. NPJ Breast Cancer 3: 34, 2017.
- Rudolph A, Song M, Brook MN, et al.: Joint associations of a polygenic risk score and environmental risk factors for breast cancer in the Breast Cancer Association Consortium. Int J Epidemiol 47 (2): 526-536, 2018.
- Shendure J: Next-generation human genetics. Genome Biol 12 (9): 408, 2011.
- Park DJ, Lesueur F, Nguyen-Dumont T, et al.: Rare mutations in XRCC2 increase the risk of breast cancer. Am J Hum Genet 90 (4): 734-9, 2012.
Clinical Management of Hereditary Breast and/or Gynecologic Cancer Syndromes
BRCA1 and BRCA2
For more information, see the Management of Cancer Risks in BRCA1/2 Carriers section in BRCA1 and BRCA2: Cancer Risks and Management.
Lynch Syndrome
As mismatch repair genes were identified as the genetic basis of Lynch syndrome, microsatellite instability was identified as a common molecular marker of mismatch repair deficiency. Approximately 15% of sporadic colorectal cancers show microsatellite instability, while up to 28% of sporadic endometrial cancers have this molecular change. Most frequently, sporadic tumors with microsatellite instability have hypermethylation of the MLH1 promoter. In Lynch syndrome–related tumors showing microsatellite instability, there is typically loss of one or more of the proteins associated with the mismatch repair genes.
Certain histopathologic features are also strongly suggestive of a microsatellite instability phenotype, including the presence of tumor infiltrating lymphocytes, peritumoral lymphocytes, undifferentiated carcinomas, and lower uterine segment tumors. Use of clinical criteria is one strategy of selection criteria for tumor testing. Computer models have also been used to predict the probability of a mismatch repair genetic variant and can be used in the absence of microsatellite instability or immunohistochemistry information. Overall, however, there is a move towards universal testing of colorectal and endometrial tumors when tumor tissue is available. (Refer to the Universal tumor testing to screen for Lynch syndrome section in Genetics of Colorectal Cancer for more information.)
References
- Vilar E, Gruber SB: Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol 7 (3): 153-62, 2010.
- Nakamura A, Osonoi T, Terauchi Y: Relationship between urinary sodium excretion and pioglitazone-induced edema. J Diabetes Investig 1 (5): 208-11, 2010.
- Balmaña J, Stockwell DH, Steyerberg EW, et al.: Prediction of MLH1 and MSH2 mutations in Lynch syndrome. JAMA 296 (12): 1469-78, 2006.
- Barnetson RA, Tenesa A, Farrington SM, et al.: Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med 354 (26): 2751-63, 2006.
- Kastrinos F, Allen JI, Stockwell DH, et al.: Development and validation of a colon cancer risk assessment tool for patients undergoing colonoscopy. Am J Gastroenterol 104 (6): 1508-18, 2009.
- Khan O, Blanco A, Conrad P, et al.: Performance of Lynch syndrome predictive models in a multi-center US referral population. Am J Gastroenterol 106 (10): 1822-7; quiz 1828, 2011.
Psychosocial Issues in Hereditary Breast and Gynecologic Cancers
Introduction
Psychosocial research in the context of cancer genetic testing helps to define psychological outcomes, interpersonal and familial effects, and cultural and community responses. This type of research also identifies behavioral factors that encourage or impede screening and other health behaviors. It can enhance decision making about risk-reduction interventions, evaluate psychosocial interventions to reduce distress and/or other negative sequelae related to risk notification and genetic testing, provide data to help resolve ethical concerns, and predict the interest in testing of various groups.
Psychosocial and screening issues related to gynecologic cancers associated with Lynch syndrome are discussed in the Psychosocial Issues in Hereditary Colon Cancer Syndromes section in Genetics of Colorectal Cancer.
Uptake of Genetic Counseling and Genetic Testing
Degree of uptake of genetic counseling and genetic testing
Comparison of uptake rates among studies in which counseling and testing were offered is challenging because of differences in methodologies, including the sampling strategy used, the recruitment setting, and testing through a research protocol with high-risk cohorts or kindreds. In a systematic review of 40 studies conducted before 2002 that had assessed genetic testing utilization, uptake rates varied widely and ranged from 25% to 96%, with an average uptake rate of 59%. Results of multivariate analysis found that BRCA1/BRCA2 genetic testing uptake was associated with having a personal or family history of breast or ovarian cancer, and with methodological features of the studies, including sampling strategies, recruitment settings, and how studies defined actual uptake versus the intention to have testing.
Other factors have been positively correlated with uptake of BRCA1/BRCA2 genetic testing, although these findings are not consistent across all studies. Psychological factors that have been positively correlated with testing uptake include greater cancer-specific distress and greater perceived risk of developing breast or ovarian cancer. Having more cancer-affected relatives also has been correlated with greater testing uptake.
Table 8 summarizes the uptake of genetic testing in clinical and research cohorts in the United States.
| Study Citation | Study Population | Sample Size (N) | Uptake of GT | Predictors Associated With Uptake of GT | Comments |
|---|---|---|---|---|---|
| GC = genetic counseling; HMO = health maintenance organization. | |||||
| aSelf-report as data source. | |||||
| bMedical records as data source. | |||||
| Schwartz et al. (2005) | Newly diagnosed and locally untreated breast cancer patients with ≥10% risk of having a BRCA1/BRCA2 pathogenic varianta | 231 | 177/231 (77%) underwent GT | Having decided on definitive local treatment. Women who were undecided on a definitive local treatment were more likely to be tested | Testing was offered free of charge |
| 34/231 (15%) had baseline interview but declined GT | |||||
| Physician recommendation for testing. Women whose physician had recommended GT were more likely to be tested | 38/177 chose to proceed with treatment before receiving test results | ||||
| 20/231 declined baseline interview | |||||
| Kieran et al. (2007) | Women who received GC between 2002 and 2004a | 250 | 88/250 (35%) underwent GT | Ability to pay for GT (entire cost or cost not covered by insurance). Nonuptake was 5.5 times more likely in women who could not afford testing | 450 women received GC for breast and ovarian cancer risk during study period. 250 women were retrospectively identified as eligible and were mailed a study questionnaire |
| 36/88 returned surveys | |||||
| Ability to recall risk estimates that were provided post-GC. Nonuptake was 15.5 times more likely in women who could not recall their risk estimates | All women had some form of insurance | ||||
| 162/250 (65%) eligible | |||||
| 65/162 returned surveys | |||||
| Susswein et al. (2008) | African American women and White women with breast cancerb | 768 | 529/768 (69%) underwent GT | Race and ethnicity. African American women were less likely to be tested than White women | Sample obtained from a clinical database. Testing was offered free of charge when it was not covered by insurance. This effect for time of diagnosis was significant in the African American subgroup but not in the White subgroup |
| African American women: 77/132 (58%) underwent GT | |||||
| Recent diagnosis. African American women who were recently diagnosed were more likely to be tested | |||||
| White women: 452/636 (71%) underwent GT | |||||
| Olaya et al. (2009) | Patients referred for GT between 2001 and 2008b | 213 | 111/213 (52%) underwent GT | Personal history of breast cancer. Having a personal history was associated with 3 times greater odds of being tested | Insurance coverage for testing was available for 91.1% (175/213) of patients. Of those who had coverage for GT, 51.4% underwent testing and 48.6% did not. Of those without coverage, 41.2% had GT and 58.9% did not |
| 102/213 (48%) declined GT | Higher level of education. Those with a high school education or less had one-third the odds of being tested, compared with those with at least some college | ||||
| Levy et al. (2010) | Women aged 20–40 y with newly diagnosed early-onset breast cancer.b | 1,474 | 446/1,474 (30%) underwent GT | Race and ethnicity. Women of Jewish ethnicity were 3 times more likely to be tested than non-Jewish White women. African American and Hispanic women were significantly less likely to receive testing than non-Jewish White women | Sample obtained from a national database of commercially insured individuals |
| Jewish women: 18/32 (56%) underwent GT | Home location. Women living in the south were more likely to be tested than women living in the northeast | ||||
| African American women: 10/82 (12%) underwent GT | Insurance type. Women with point-of-service plans were more likely to be tested than women with HMO plans | ||||
| Recent diagnosis. Women diagnosed in 2007 were 3.8 times more likely to be tested than women diagnosed in 2004 | |||||
Several studies conducted in non-U.S. settings have examined the uptake of genetic testing. In studies examining the uptake of testing among at-risk relatives of carriers of BRCA1/BRCA2 pathogenic variants, uptake rates have averaged below 50% (range, 36%–48%), with higher uptake reported among female relatives than in male relatives. Other factors associated with higher uptake of testing were not consistently reported among studies but have most commonly included being a parent and wanting to learn information about a child’s risk.
Factors influencing uptake of genetic counseling and genetic testing
In reviews that have examined the cumulative evidence concerning the predictors of uptake of BRCA1/BRCA2 genetic testing, important predictors of testing uptake include older age, Ashkenazi Jewish (AJ) heritage, unmarried status, a personal history of breast cancer, and a family history of breast cancer. Studies recruiting participants in hospital settings had significantly higher recruitment rates than did studies recruiting participants in community settings. Studies that required an immediate decision to test, rather than allowing delayed decision making, tended to report higher uptake rates. However, there is evidence that women diagnosed with breast cancer are equally satisfied with genetic counseling (including information received and strength and timing of physician recommendations for counseling), whether they received genetic counseling before or after their definitive surgery for breast cancer. Another review found that uptake of genetic testing for BRCA1/BRCA2 pathogenic variants was related to psychological factors (e.g., anxiety about breast cancer and perceived risk of breast cancer) and demographic and medical factors (e.g., history of breast cancer or ovarian cancer, presence of children, and higher number of affectedfirst-degree relatives [FDRs]). Family members with a known BRCA1/BRCA2 pathogenic variant were more likely to pursue testing; those with more extensive knowledge of BRCA1/BRCA2 testing, heightened risk perceptions, beliefs that mammography would promote health benefit, and high intentions to undergo testing were more likely to follow through with testing.
In a review of racial and ethnic differences that affect the uptake of BRCA1/BRCA2 testing, intention to undergo genetic testing in African American women was related to having at least one FDR with breast cancer or ovarian cancer, higher perceived risk of being a carrier, and less anticipatory guilt about the possibility of being a gene carrier. A systematic review found that certain racial and ethnic minority groups, including African American and Hispanic individuals, had more negative views and greater concerns about genetic counseling and testing when compared with White individuals. African American and Hispanic individuals were more likely to believe genetic testing could be used to show their ethnic group was inferior to other groups. Additionally, African American and Hispanic individuals were found to have low awareness and knowledge about the importance of genetics in cancer, BRCA status, and genetic testing.
Reasons cited for following through with testing included a desire to learn about a child's risk, to feel relief from uncertainty, to inform screening or risk-reducing surgery decisions, and to inform important life decisions such as marriage and childbearing. Among African American women, the most important reason for testing included motivation to help other relatives decide on genetic testing.
Physician recommendation may be another motivator for testing. In a retrospective study of 335 women considering genetic testing, 77% reported that they wanted the opinion of a genetics physician about whether they should be tested, and 49% wanted the opinion of their primary care provider. However, there is some evidence of referral bias favoring those with a maternal family history of breast cancer or ovarian cancer. In a Canadian retrospective review of 315 patients, those with a maternal family history of breast cancer or ovarian cancer were 4.9 times (95% confidence interval, 3.6–6.7) more likely to be referred for a cancer genetics consultation by their physician than were those with a paternal family history (P< .001). Studies have found that physicians may not adequately assess paternal family history or may underestimate the significance of a paternal family history for genetic risk. Other studies have shown that physician referral of patients who meet U.S. Preventive Services Task Force guidelines for BRCA genetic counseling has been suboptimal.
The uptake of BRCA testing to inform surgical treatment decisions when offered appears to be high in research cohorts; however, findings from other studies suggest that testing is underutilized in clinical practice to inform breast cancer treatment decisions. Barriers to the use of BRCA testing to inform surgical treatment decisions, including lack of physician referral of newly diagnosed patients for genetic counseling, type of insurance coverage (such as Medicare or Medicaid), and challenges in the timing and coordination of testing, have been reported. In a randomized trial that provided proactive rapid genetic counseling (delivery of genetic counseling prior to surgery) compared with usual care for patients with newly diagnosed breast cancer, results suggested that although genetic counseling uptake was higher in the intervention arm, this did not translate into higher rates of genetic testing, receipt of results before surgery, or bilateral mastectomy decisions.
Insurance coverage
Insurance coverage is an important consideration for individuals deciding whether to undergo genetic testing. (Refer to the Insurance coverage section in the PDQ summary on Cancer Genetics Risk Assessment and Counseling for more information.)
Uptake of genetic counseling and genetic testing in diverse populations
Degree of uptake of genetic counseling and genetic testing in diverse populations
There are limited data on uptake of genetic counseling and testing among non-White populations, and further research will be needed to define factors influencing uptake in these populations. The uptake of BRCA testing appears to vary across some racial and ethnic groups. A few studies have compared uptake rates between African American and White women. In a case-control study of women who had been seen in a university-based primary care system, African American women with family histories of breast cancer or ovarian cancer were less likely to undergo BRCA1/BRCA2 testing than were White women who had similar histories. In another study among breast cancer patients who were counseled about BRCA1/BRCA2 risk in a clinical setting, lower uptake was reported among African American women than among White women.
Notably, the racial differences observed in these studies do not appear to be explained by factors related to cost, access to care, risk factors for carrying a BRCA1 or BRCA2 pathogenic variant, or differences in psychosocial factors, including risk perceptions, worry, or attitudes toward testing.
Factors influencing uptake of genetic counseling and genetic testing in diverse populations
Several studies have examined uptake or “acceptance” of BRCA testing among African American individuals enrolled in genetic research programs. Among study enrollees from an African American kindred in Utah, 83% underwent BRCA1 testing. Age, perceived risk of being a carrier, and more extensive cancer knowledge predicted testing acceptance. Another study that recruited African American women through physician and community referrals reported a BRCA1/BRCA2 testing acceptance rate of 22%. Predictors of test acceptance included having a higher probability of having a pathogenic variant, being married, and being less certain about one’s cancer risk. Finally, a third study that recruited at-risk African American women from an urban cancer screening clinic found that acceptors of BRCA testing were more knowledgeable about breast cancer genetics and perceived fewer barriers to testing, including negative emotional reactions, stigmatization concerns, and family-related guilt. While these are independent predictors of genetic testing uptake, they do not explain the disparities in testing uptake across different ethnic groups. What may explain these differences are several attitudes and beliefs held about testing by individuals from diverse populations.
Work examining attitudes toward breast cancer genetic testing in Latino and African American populations indicates limited knowledge and awareness about testing but a generally receptive view once they are informed; in comparison with White populations, Latino and African American populations have relatively more concerns about testing.
For example, in a qualitative study with 51 Latino individuals unselected for risk status, important findings included the fact that participants were highly interested in genetic testing for inherited cancer susceptibility, despite very limited knowledge about genetics. One important barrier involved secrecy or embarrassment about family discussions of cancer and genetics, which could be addressed in intervention strategies. Another qualitative study with 54 Latina women at risk of hereditary breast cancer showed that knowledge about BRCA1/BRCA2 counseling was low, although the women were interested in learning more about counseling to gain risk information for family members. Barriers to counseling included life demands, cost, and language issues.
A telephone survey of 314 patients from an inner-city network of Pittsburgh, Pennsylvania, health centers, 50% of whom were African American, found that most participants (57%) (both African American and White participants) felt that genetic testing to evaluate disease risk was a good idea; however, more African American participants than White participants thought that genetic testing would lead to racial discrimination (37% vs. 22%, respectively) and that genetics research was unethical and tampered with nature (20% vs. 11%, respectively). Finally, in a study of 222 women in Savannah, Georgia, where most had neither a personal history (70%) nor a family history (60%) of breast cancer, African American women (who comprised 26% of the sample) were less likely to be aware of breast cancer genes and genetic testing. Awareness was also related to higher income, higher education level, and having a family breast cancer history. However, 74% of the entire sample expressed willingness to be tested for breast cancer susceptibility.
In a sample of 146 African American women meeting criteria for BRCA1/BRCA2 pathogenic variant testing, women born outside the United States reported higher levels of anticipated negative emotional reactions (e.g., fear, hopelessness, and lack of confidence that they could emotionally handle testing). Higher levels of breast cancer–specific distress were associated with anticipated negative emotional reactions, confidentiality concerns, and anticipated guilt regarding the family impact of breast cancer genetic testing. A future orientation (e.g., "I often think about how my actions today will affect my health when I am older") was associated with overall perceived benefits of breast cancer genetic testing in this population (n = 140); however, future orientation was also found to be positively associated with family-related cons of testing, including family guilt and worry regarding the impact of testing on the family.
There are racial differences in provider discussion and patient uptake of genetic testing for variants in BRCA1/BRCA2. A study of women aged 18 to 64 years and diagnosed with invasive breast cancer between 2007 and 2009 found that, even after adjusting for pathogenic variant risk, African American women were less likely to report having received a physician recommendation for genetic testing. There was no difference across all races in concerns that BRCA1/BRCA2 testing was too expensive and only minimal differences in testing attitudes or insurance concerns were found, none of which influenced testing uptake. A study of breast or ovarian cancer survivors (N = 50) eligible for BRCA1/BRCA2 genetic testing found that 48% were referred for genetic counseling and testing and/or had undergone genetic testing. Individuals with higher breast cancer genetics knowledge and higher self-efficacy were more likely to have engaged in genetic counseling and testing. In a study of women with invasive breast cancer diagnosed before age 50 years between 2009 and 2012 who were identified through the Florida Cancer Data System state registry and eligible for BRCA1/BRCA2 genetic testing on the basis of existing guidelines, African American individuals were less likely to report a discussion with their health care provider and undergo genetic testing. The same study found similar overall testing rates in Hispanic (61%) and non-Hispanic (65%) White individuals. However, testing rates were lower among Hispanic individuals who spoke primarily Spanish at home (50% Spanish speaking vs. 69% English speaking; P = .0009), and in general, Hispanic individuals were less likely to have been referred for genetic testing. However, this finding is not consistent across all studies. In a study of women aged 20 to 79 years with ductal carcinoma in situ or invasive breast cancer identified through the Surveillance, Epidemiology, and End Results (SEER) registry in Georgia and Los Angeles County, all eligible for BRCA1/BRCA2 genetic testing on the basis of existing guidelines, no ethnic differences were detected in receipt of genetic counseling or physician-directed discussion about genetic testing.
Factors associated with declining genetic counseling and testing
There is evidence that primary reasons for declining testing involves being childless, which reduces any family motivations for testing; and concerns about the negative ramifications of testing, including difficulty retaining insurance or concerns about personal health.
Limited data are available about the characteristics of at-risk individuals who decline to be tested or have never been tested. It is difficult to access samples of test decliners because they may be reluctant to participate in research studies. Studies of genetic testing uptake are difficult to compare because people may decline at different points and with different amounts of pretest education and counseling. One study found that 43% of affected and unaffected individuals from hereditary breast/ovarian cancer families who completed a baseline interview regarding testing declined to be tested. Most individuals who declined testing chose not to participate in educational sessions. Decliners were more likely to be male and be unmarried and have fewer relatives with breast cancer. Decliners who had high levels of cancer-related stress had higher levels of depression. Decliners lost to follow-up were significantly more likely to be affected with cancer.
Another study looked at a small number (n = 13) of women decliners who carried a 25% to 50% probability of harboring a BRCA pathogenic variant; these nontested women were more likely to be childless and to have higher levels of education. This study showed that most women decided not to undergo the test after serious deliberation about the risks and benefits. Satisfaction with frequent surveillance was given as one reason for nontesting by most of these women. Other reasons for declining included having no children and becoming acquainted with breast/ovarian cancer in the family relatively early in their lives.
A third study evaluated characteristics of 34 individuals who declined BRCA1/BRCA2 testing in a large multicenter study in the United Kingdom. Decliners were younger than a national sample of test acceptors, and female decliners had lower mean scores on a measure of cancer worry. Although 78% of test decliners/deferrers felt that their health was at risk, they reported that learning about their BRCA1/BRCA2 pathogenic variant status would cause them to worry about the following:
- Their children's health (76%).
- Their life insurance (60%).
- Their own health (56%).
- Loss of their job (5%).
- Receiving less screening if they did not carry a BRCA1/BRCA2 pathogenic variant (62%).
Apprehension about the impact of the test result was a more important factor in the decision to decline testing than were concrete burdens such as time required to travel to a genetics clinic and time spent away from work, family, and social obligations. In 15% (n = 31) of individuals from 13 hereditary breast and ovarian cancer (HBOC) families who underwent genetic education and counseling and declined testing for a documented pathogenic variant in the family, positive changes in family relationships were reported—specifically, greater expressiveness and cohesion—compared with those who pursued testing.
Genetic counseling and testing in children
Testing for BRCA1/BRCA2 pathogenic variants has been almost universally limited to adults older than 18 years. The risks of testing children for adult-onset disorders, such as breast and ovarian cancers, as inferred from developmental data on children’s medical understanding and ability to provide informed consent, have been outlined in several reports.
Studies suggest that individuals who have undergone BRCA1/BRCA2 genetic testing or who are adult offspring of individuals who have had testing are generally not in favor of testing minors. Although the data are limited, research suggests that males, pathogenic variant noncarriers, and those whose mothers did not have personal histories of breast cancer may be more likely to favor genetic testing in minors in general. Of those who had minor children at the time the study was conducted, only 17% stated a preference for having their own children tested. Concerns regarding testing of minors included psychological risks and insufficient maturity. Potential benefits included the ability to influence health behaviors.
No data exist on the testing of children for BRCA1/BRCA2 pathogenic variants, although some researchers believe it is necessary to test the validity of assumptions underlying the general prohibition of testing children for genetic variants associated with breast and ovarian cancers and other adult-onset diseases. In one study, 20 children (aged 11–17 y) of a selected group of mothers undergoing genetic testing (80% of whom previously had breast cancer and all of whom had discussed BRCA1/BRCA2 testing with their children) completed self-report questionnaires on their health beliefs and attitudes toward cancer, feelings related to cancer, and behavioral problems. Ninety percent of children thought they would want cancer risk information as adults; half worried about themselves or a family member developing cancer. There was no evidence of emotional distress or behavioral problems.
What People Bring to Genetic Testing: Impact of Risk Perception, Health Beliefs, and Personality Characteristics
The emerging literature in this area suggests that risk perceptions, health beliefs, psychological status, and personality characteristics are important factors in decision making about breast/ovarian cancer genetic testing. Many women presenting at academic centers for BRCA1/BRCA2 testing arrive with a strong belief that they have a pathogenic variant, having decided they want genetic testing, but possessing little information about the risks or limitations of testing. Most mean scores of psychological functioning at baseline for subjects in genetic counseling studies were within normal limits. Nonetheless, a subset of subjects in many genetic counseling studies present with elevated anxiety, depression, or cancer worry. Identification of these individuals is essential to prevent adverse outcomes. In a study of 205 women pursuing genetic counseling, interactions among cancer worry, breast cancer risk perception, and perceived severity of having a breast cancer genetic variant were found such that those with high worry, high breast cancer risk perception, and low perceived severity were twice as likely to follow through with BRCA1/BRCA2 testing than others.
A general tendency to overestimate inherited risk of breast and ovarian cancer has been noted in at-risk populations, in cancer patients, in spouses of breast and ovarian cancer patients, and among women in the general population. but underestimation of breast cancer risk in higher-risk and average-risk women also has been reported. This overestimation may encourage a belief that BRCA1/BRCA2 genetic testing will be more informative than it is currently thought to be. Some evidence exists that even counseling does not dissuade women at low to moderate risk from the belief that BRCA1 testing could be valuable. Overestimation of both breast and ovarian cancer risk has been associated with nonadherence to physician-recommended screening practices. A meta-analysis of 12 studies of outcomes of genetic counseling for breast/ovarian cancer showed that counseling improved the accuracy of risk perception.
Women appear to be the prime communicators within families about the family history of breast cancer. Higher numbers of maternal versus paternal transmission cases are reported, likely due to family communication patterns, to the misconception that breast cancer risk can only be transmitted through the mother, and to the greater difficulty in recognizing paternal family histories because of the need to identify more distant relatives with cancer. In an analysis of 2,505 women participating in the Family Healthware Impact Trial, not only was evidence of underreporting of paternal family history identified, but also women reported a lower level of perceived breast cancer risk with a paternal versus maternal breast cancer family history. Physicians and counselors taking a family history are encouraged to elicit paternal and maternal family histories of breast, ovarian, or other associated cancers.
The accuracy of reported family history of breast or ovarian cancer varies; some studies found levels of accuracy above 90%, with others finding more errors in the reporting of cancer in second-degree relatives (SDRs) or more distant relatives or in age of onset of cancer. Less accuracy has been found in the reporting of cancers other than breast cancer. Ovarian cancer history was reported with 60% accuracy in one study compared with 83% accuracy in breast cancer history. Providers should be aware that there are a few published cases of Munchausen syndrome in reporting of false family breast cancer history. Much more common is erroneous reporting of family cancer history due to unintentional errors or gaps in knowledge, related in some cases to the early death of potential maternal informants about cancer family history. (Refer to the Documenting the Family History section in Cancer Genetics Risk Assessment and Counseling for more information.)
Targeted written, video, CD-ROM, interactive computer programs and websites, and culturally targeted educational materials may be effective and efficient methods of increasing knowledge about the pros and cons of genetic testing. Such supplemental materials may allow more efficient use of the time allotted for pretest education and counseling by genetics and primary care providers and may discourage individuals without appropriate indication of risk from seeking genetic testing.
Genetic Counseling for Hereditary Predisposition to Breast Cancer
Counseling for breast cancer risk typically involves individuals with family histories that are potentially attributable to BRCA1 or BRCA2. It also, however, may include individuals with family histories of Li-Fraumeni syndrome, ataxia-telangiectasia, Cowden syndrome, or Peutz-Jeghers syndrome. (Refer to the High-Penetrance Breast and/or Gynecologic Cancer Susceptibility Genes section of this summary for more information.)
Management strategies for carriers may involve decisions about the nature, frequency, and timing of screening and surveillance procedures, chemoprevention, risk-reducing surgery, and use of hormone replacement therapy (HRT). The utilization of breast conservation and radiation as cancer therapy for women who are carriers may be influenced by knowledge of pathogenic variant status. (Refer to the Management of Cancer Risks in BRCA1/2 Carriers section in BRCA1 and BRCA2: Cancer Risks and Management for more information.)
Counseling also includes consideration of related psychosocial concerns and discussion of planned family communication and the responsibility to warn other family members about the possibility of having an increased risk of breast, ovarian, and other cancers. Data suggest that individual responses to being tested as adults are influenced by the results status of other family members. Management of anxiety and distress are important not only as quality-of-life factors, but also because high anxiety may interfere with the understanding and integration of complex genetic and medical information and adherence to screening. Formal, objective evaluation of these outcomes are well documented. (Refer to the Emotional Outcomes and Behavioral Outcomes sections of this summary for more information.)
Published descriptions of counseling programs for BRCA1 (and subsequently for BRCA2) testing include strategies for gathering a family history, assessing eligibility for testing, communicating the considerable volume of relevant information about breast/ovarian cancer genetics and associated medical and psychosocial risks and benefits, and discussion of specialized ethical considerations about confidentiality and family communication. Participant distress, intrusive thoughts about cancer, coping style, and social support were assessed in many prospective testing candidates. The psychosocial outcomes evaluated in these programs have included changes in knowledge about the genetics of breast/ovarian cancer after counseling, risk comprehension, psychological adjustment, family and social functioning, and reproductive and health behaviors. A Dutch study of communication processes and satisfaction levels of consultands going through cancer genetic counseling for inherited cancer syndromes indicated that asking more medical questions (by the counselor), providing more psychosocial information, and longer eye contact by the counselor were associated with lower satisfaction levels. The provision of medical information by the counselor was most highly related to satisfaction and perception that needs have been fulfilled.
Many of the psychosocial outcome studies involve specialized, highly selected research populations, some of which were utilized to map and clone BRCA1 and BRCA2. One such example is K2082, an extensively studied kindred of more than 800 members of a Utah Mormon family in which a BRCA1 pathogenic variant accounts for the observed increased rates of breast and ovarian cancer. A study of the understanding that members of this kindred have about breast/ovarian cancer genetics found that, even in breast cancer research populations, there was incomplete knowledge about associated risks of colon and prostate cancer, the existence of options for risk-reducing mastectomy (RRM) and risk-reducing salpingo-oophorectomy (RRSO), and the complexity of existing psychosocial risks. A meta-analysis of 21 studies found that genetic counseling was effective in increasing knowledge and improved the accuracy of perceived risk. Genetic counseling did not have a statistically significant long-term impact on affective outcomes including anxiety, distress, or cancer-specific worry and the behavioral outcome of cancer surveillance activities. These prospective studies, however, were characterized by a heterogeneity of measures of cancer-specific worry and inconsistent findings in effects of change from baseline.
Emotional Outcomes
Although there were initial concerns about the possibility of adverse emotional consequences from BRCA testing, most studies conducted over the years have shown low levels of psychological distress among both carriers and noncarriers, particularly over the longer term. In a meta-analysis examining cancer-specific distress over short (0–4 weeks), moderate (5–24 weeks), and long (25–52 weeks) periods of time since the receipt of testing results, carriers were found to demonstrate increased levels of distress shortly after receiving results, with levels returning to baseline within moderate and long periods of time. In contrast, noncarriers and those with inconclusive results showed reduced levels of distress over time. Psychological distress patterns were found to vary as a function of several factors, including the cancer history of the individual and the country within which the study was conducted. Carriers with a personal history of cancer experienced small decreases in distress over time, whereas no changes were observed among carriers without a personal history of cancer. Among individuals with inconclusive results, greater decreases in distress were observed among those without a cancer history than among those with a cancer history. Among noncarriers, those in the United States experienced significantly greater decreases in psychological distress than noncarriers from Europe and Australia. A study conducted in Austria noted that certain subgroups of consultands experienced greater distress, including those who were older, had a more recent cancer diagnosis, or those who had received counseling but declined BRCA testing.
Several studies have reported on emotional outcomes over longer follow-up periods (i.e., greater than 12 months after disclosure) than those reported in the meta-analysis described above. In a U.K. study, cancer-related worry did not differ between carriers and noncarriers at 3 years of follow-up. Two U.S.-based studies published since the meta-analytic review have reported similar findings among women who were surveyed more than 3 years after receipt of BRCA test results. In a cross-sectional study, 167 women who were surveyed more than 4 years after receiving BRCA test results reported low levels of genetic testing–specific concerns, as measured using the Multidimensional Impact of Cancer Risk Assessment Scale. In multivariate regression models, carriers of pathogenic variants were significantly more likely to experience distress than were noncarriers. In a second study, 464 women were followed prospectively for a median of 5 years (range, 3.4–9.1 y) after testing. Among both affected and unaffected participants, BRCA carriers reported significantly higher levels of distress, uncertainty (affected only), perceived stress (affected only), and lower positive testing experiences (unaffected only) than women who received negative results for a known pathogenic variant in the family. Although both studies reported greater distress among BRCA carriers than among noncarriers, the level of distress was not reflective of clinically significant dysfunction.
Although most studies have reported that a positive BRCA test result has a relatively minimal impact on psychological distress, many of these studies were conducted among families with a strong family history of breast or ovarian cancer who underwent extensive pretest genetic counseling. Therefore, emotional responses may not generalize to individuals who test under different contexts. For example, individuals who are tested with population BRCA screening may not have a family history of cancer. Although pretest genetic counseling is recommended, this is not always done when genetic testing is ordered by nongenetic providers or directly through commercial companies.
For example, in a Canadian study of 2,080 Jewish women who participated in a population-based genetic screening study to test for three BRCA pathogenic variants common in families of Jewish heritage, women were not offered in-person genetic counseling but were given a pamphlet on genetic testing for BRCA1/BRCA2 before they provided a DNA sample. One year after genetic testing, women who were positive for a pathogenic variant (n = 18) showed significant increases in cancer-specific distress, whereas no changes in distress were observed among women who were negative for a pathogenic variant. The mean distress score on the Impact of Event Scale for the 18 women with a known pathogenic variant was 25.3 (range, 2–51); 10 of 18 women (56%) scored within moderate (26–43) (n = 7) or severe (44+) (n = 3) ranges. It is unclear from this study whether the increase in distress observed at 1 year of follow-up was due to the lack of in-person genetic counseling, or whether the lower levels of distress at baseline observed were because the women in the study were low risk but eligible for testing because of their ancestry. A follow-up study with this cohort found that distress decreased between 1 to 2 years after testing and that changes in distress varied by risk-reduction options undertaken by carriers. Specifically, those who had undergone RRM or risk-reducing oophorectomy experienced significant decreases in distress compared with those who did not have either surgery. Another smaller qualitative study also supports these findings.
Similarly, the impact of direct-to-consumer (DTC) BRCA testing through commercial companies requires further evaluation. Case studies have reported adverse emotional responses after receipt of a positive BRCA result from DTC genetic testing, suggesting the need for further evaluation of the emotional outcomes of women undergoing genetic testing through commercial companies. Only one study, conducted by a commercial company, has attempted to evaluate the impact of BRCA testing in this context. A total of 32 individuals (16 women and 16 men) who tested positive for one of three BRCAfounder pathogenic variants common in AJs completed semi-structured interviews. None of the carriers reported extreme anxiety, although some experienced moderate anxiety (13%) or initial disappointment and anxiety that dissipated over time (28%). These findings should be interpreted with caution given that only 24% (32 of 136) of invited carriers of BRCA pathogenic variants participated in the study, raising concerns about selection bias.
Despite evidence of a short-term increase in distress after the receipt of genetic testing results, any adverse responses to a positive carrier status dissipate within 12 months. Additional research is needed to examine emotional outcomes for those who are not provided genetic counseling before testing.
Emotional outcomes in newly diagnosed breast cancer patients
It is increasingly common for women with breast cancer to pursue genetic counseling and testing at the time of diagnosis to assist with treatment decision making. (Refer to the Benefits of offering genetic testing at the time of cancer diagnosis section for more information.) Given that women with new breast cancer diagnoses are likely to experience some distress, concerns have been raised about the potential for additional adverse psychological implications of rapid genetic counseling and testing (RGT) between diagnosis and surgery. However, data from several studies, including a large observational study and a randomized trial, provided evidence indicating that there were no adverse psychological effects associated with RGT. For example, an observational study analyzed 1,007 with newly diagnosed breast cancers from four Canadian academic centers. These patients underwent RGT and adverse psychological effects were not noted at one week or one year posttesting. There were no differences in anxiety, distress, or depression between BRCA carriers (6% of the study population) and those with uninformative results in the short-term. However, after one year, BRCA carriers had significantly lower depression scores when compared with patients who had uninformative, negative results. Depression scores were within the normal range for both groups, and these scores did not reach depression levels that were clinically significant. Factors associated with lower levels of psychological functioning included lower educational levels, not having family histories of cancer, and undergoing bilateral mastectomies. Of note, women who had a variant of uncertain significance in the BRCA1/2 genes and a bilateral mastectomy had higher depression scores than BRCA carriers. One randomized controlled trial found that patients undergoing rapid genetic counseling and testing felt more actively involved in treatment decision making than those receiving standard care.
Family Effects
Family communication about genetic testing and hereditary risk
Family communication about genetic testing for cancer susceptibility, and specifically about the results of BRCA1/BRCA2 genetic testing, is complex. Age and gender appear to be important variables in the disclosure of genetic test results. Studies have documented that older women were more likely to disclose genetic test results (especially to their daughters) than younger women. Female carriers are more likely to disclose their genetic test results to other family members than are male carriers. Among males, noncarriers were more likely than carriers to tell their sisters and children the results of their tests. BRCA1/BRCA2 carriers who disclosed their results to sisters had a slight decrease in psychological distress, compared with a slight increase in distress for carriers who chose not to tell their sisters. One study found that men reported greater difficulty disclosing a known pathogenic variant to family members than women (90% vs. 70%).
Family communication of BRCA1/BRCA2 test results to relatives is another factor affecting participation in testing. There have been more studies of communication with FDRs and SDRs than with more distant family members. Studies have investigated the process and content of communication among sisters about BRCA1/BRCA2 test results. Study results suggest that both carriers of pathogenic variants and women with uninformative results communicate with sisters to provide them with genetic risk information. Similar findings were reported in women with uninformative results disclosing test results to their daughters.
Among relatives with whom genetic test results were not discussed, the most important reason given was that the affected women were not close to their relatives or had a poor relationship with them. Studies found that women with a BRCA pathogenic variant more often shared their results with their mother and adult sisters and daughters than with their father and adult brothers and sons.
A study that evaluated communication of test results to FDRs at 4 months postdisclosure found that participants were more likely to inform brothers of their results if the BRCA pathogenic variant was inherited through the paternal line. Another study found that disclosure was limited mainly to FDRs, and dissemination of information to distant relatives was problematic. Age was a significant factor in informing distant relatives with younger patients being more willing to communicate their genetic test result. Additionally, one study found that lower genetic worry, higher interest in genomic information, carrying a BRCA1 or BRCA2 pathogenic variant, or having never been married was associated with communication to more family members. In contrast, a longer time interval since diagnosis was associated with communication to fewer family members.
A few in-depth qualitative studies have looked at issues associated with family communication about genetic testing. Although the findings from these studies may not be generalizable to the larger population of at-risk individuals, they illustrate the complexity of issues involved in conveying hereditary cancer risk information in families. On the basis of 15 interviews conducted with women attending a familial cancer genetics clinic, the authors concluded that while women felt a sense of duty to discuss genetic testing with their relatives, they also experienced conflicting feelings of uncertainty, respect, and isolation. Decisions about whom in the family to inform and how to inform them about hereditary cancer and genetic testing may be influenced by tensions between women's need to fulfill social roles and their responsibilities toward themselves and others. Another qualitative study of 21 women who attended a familial breast and ovarian cancer genetics clinic suggested that some women may find it difficult to communicate about inherited cancer risk with their partners and with certain relatives, especially brothers, because of their own fears and worries about cancer. This study also suggested that how genetic risk information is shared within families may depend on the existing norms for communicating about cancer in general. For example, family members may be generally open to sharing information about cancer with each other, may selectively avoid discussing cancer information with certain family members to protect themselves or other relatives from negative emotional reactions, or may ask a specific relative to act as an intermediary to disclosure of information to other family members. The potential importance of individuals outside the family, such as friends, as both confidantes about inherited cancer risk information and as sources of support for coping with this information was also noted in the study.
A study of 31 mothers with a documented BRCA pathogenic variant explored patterns of dissemination to children. Of those who chose to disclose test results to their children, age of offspring was the most important factor. Fifty percent of the children who were told were aged 20 to 29 years and slightly more than 25% of the children were aged 19 years or younger. Sons and daughters were notified in equal numbers. More than 70% of mothers informed their children within a week of learning their test result. Ninety-three percent of mothers who chose not to share their results with their children indicated that it was because their children were too young. These findings were consistent with three other studies showing that children younger than 13 years were less likely to be informed about test results compared with older children. Another study of 187 mothers undergoing BRCA1/BRCA2 testing evaluated their need for resources to prepare for a facilitated conversation about sharing their BRCA1/BRCA2 testing results with their children. Seventy-eight percent of mothers were interested in three or more resources, including literature (93%), family counseling (86%), talk to prior participants (79%), and support groups (54%).
A longitudinal study of 153 women self-referred for genetic testing for BRCA1 and BRCA2 pathogenic variants and 118 of their partners evaluated communication about genetic testing and distress before testing and at 6 months posttesting. The study found that most couples discussed the decision to undergo testing (98%), most test participants felt their partners were supportive, and most women disclosed test results to their partners (97%, n = 148). Test participants who felt their partners were supportive during pretest discussions experienced less distress after disclosure, and partners who felt more comfortable sharing concerns with test participants pretest experienced less distress after disclosure. Six-month follow-up revealed that 22% of participants felt the need to talk about the testing experience with their partners in the week before the interview. Most participants (72%, n = 107) reported comfort in sharing concerns with their partners, and 5% (n = 7) reported relationship strain as a result of genetic testing. In couples in which the woman had a positive genetic test result, more relationship strain, more protective buffering of their partners, and more discussion of related concerns were reported than in couples in which the woman had a true-negative or uninformative result.
A study of 561 FDRs of women who had undergone BRCA1/BRCA2 genetic testing found that 22% of FDRs did not recall being informed of the genetic test results despite the women reporting that the results had been shared. Men were less likely to recall receiving the results (P > .001). Of those with recall about receiving the test results, 10.5% of FDRs did not recall the findings. For those with recall of the results, 17.9% of FDRs had an interpretation that was discordant with the correct results. Accuracy of test results recall was greater for informative test results (those that were either true positive or true negative) (P = .029). However, regardless of the test results, FDRs perceived the cancer risk to be higher before they learned of the findings than after (74% and 53% of FDRs reported that they believed their risk for cancer was greater than average before and after hearing test results, respectively).
There is a small but growing body of literature regarding psychological effects in men who have a family history of breast cancer and who are considering or have had BRCA testing. A qualitative study of 22 men from 16 high-risk families in Ireland revealed that more men in the study with daughters were tested than men without daughters. These men reported little communication with relatives about the illness, with some men reporting being excluded from discussion about cancer among female family members. Some men in the study also reported actively avoiding open discussion with daughters and other relatives. In contrast, a study of 59 men testing positive for a BRCA1/BRCA2 pathogenic variant found that most men participated in family discussions about breast and/or ovarian cancer. However, fewer than half of the men participated in family discussions about risk-reducing surgery. The main reason given for having BRCA testing was concern for their children and a need for certainty about whether they could have transmitted the pathogenic variant to their children. In this study, 79% of participating men had at least one daughter. Most of these men described how their relationships had been strengthened after receipt of BRCA results, helping communication in the family and greater understanding. Men in both studies expressed fears of developing cancer themselves. Irish men especially reported fear of cancer in sexual organs.
Family functioning
One study assessed 212 individuals from 13 families with HBOC who received genetic counseling and were offered BRCA1/BRCA2 testing for documented pathogenic variants in the family. Individuals who were not tested were found 6 to 9 months later to have significantly greater increases in family expressiveness and cohesiveness compared with those who were tested. Individuals who were randomly assigned to a client-centered versus problem-solving genetic counseling intervention had a significantly greater reduction in conflict, regardless of the test decision.
Partners of high-risk women
Many studies have looked at the psychological effects in women of having a high risk of developing cancer, either on the basis of carrying a BRCA1/BRCA2 pathogenic variant or having a strong family history of cancer. Some studies have also examined the effects on the partners of such women.
A Canadian study assessed 59 spouses of women found to have a BRCA1/BRCA2 pathogenic variant. All were supportive of their spouses’ decision to undergo genetic testing and 17% wished they had been more involved in the genetic testing process. Spouses who reported that genetic testing had no impact on their relationship had long-term relationships (mean duration 27 years). Forty-six percent of spouses reported that their major concern was of their partner dying of cancer. Nineteen percent were concerned their spouse would develop cancer and 14% were concerned their children would also be carriers of BRCA1/BRCA2 pathogenic variants.
In a U.S. study, 118 partners of women who underwent genetic testing for pathogenic variants in BRCA1 and BRCA2 completed a survey before testing and then again 6 months after result disclosure. At 6 months, only 10 partners reported that they had not been told of the test result. Ninety-one percent reported that the testing had not caused strain on their relationship. Partners who were comfortable sharing concerns before testing experienced less distress after testing. Protective buffering was not found to impact distress levels of partners.
An Australian study of 95 unaffected women at high risk of developing breast and/or ovarian cancer (13 carriers of pathogenic variants and 82 with unknown variant status) and their partners showed that although the majority of male partners had distress levels comparable to a normative population sample, 10% had significant levels of distress that indicated the need for further clinical intervention. Men with a high monitoring coping style and greater perceived breast cancer risk for their wives reported higher levels of distress. Open communication between the men and their partners and the occurrence of a cancer-related event in the wife’s family in the last year were associated with lower distress levels. When men were asked what kind of information and support they would like for themselves and their partners, 57.9% reported that they would like more information about breast and ovarian cancer, and 32.6% said they would like more support in dealing with their partner's risk. Twenty-five percent of men had suggestions on how to improve services for partners of high-risk women, including strategies on how to best support their partner, greater encouragement from health care professionals to attend appointments, and meeting with other partners.
A review of this literature reported that the BRCA testing process may be distressing for male partners, particularly for those with spouses identified as carriers. Male partner distress appears to be associated with their beliefs about the woman’s breast cancer risk, lack of couple communication, and feelings of alienation from the testing process.
At-risk males
A review of the literature on the experiences of males in families with a known BRCA1 and BRCA2 pathogenic variant reported that while the data are limited, men from variant-positive families are less likely than females to participate in communication regarding genetics at every level, including the counseling and testing process. Men are less likely to be informed of genetic test results received by female relatives, and most men from these families do not pursue their own genetic testing.
A study of Dutch men at increased risk of having inherited a BRCA1 pathogenic variant reported a tendency for the men to deny or minimize the emotional effects of their risk status, and to focus on medical implications for their female relatives. Men in these families, however, also reported considerable distress in relation to their female relatives. In another study of male psychological functioning during breast cancer testing, 28 men belonging to 18 different high-risk families (with a 25% or 50% risk of having inherited a BRCA1/BRCA2 pathogenic variant) participated. The study purpose was to analyze distress in males at risk of carrying a BRCA1/BRCA2 pathogenic variant who applied for genetic testing. Of the men studied, most had low pretest distress; scores were lowest for men who were optimistic or who did not have daughters. Most carriers of pathogenic variants had normal levels of anxiety and depression and reported no guilt, though some anticipated increased distress and feelings of responsibility if their daughters developed breast or ovarian cancer. None of the noncarriers reported feeling guilty. In one study, adherence to recommended screening guidelines after testing was analyzed. In this study, more than half of male carriers of pathogenic variants did not adhere to the screening guidelines recommended after disclosure of genetic test results. These findings are consistent with those for female carriers of BRCA1/BRCA2 pathogenic variants.
A multicenter U.K. cohort study examined prospective outcomes of BRCA1/BRCA2 testing in 193 individuals, of which 20% were men aged 28 to 86 years. Men’s distress levels were low, did not differ among carriers and noncarriers, and did not change from baseline (before genetic testing) to the 3-year follow-up. Twenty-two percent of male carriers of pathogenic variants received colorectal cancer screening and 44% received prostate cancer screening; however, it is unclear whether men in this study were following age-appropriate screening guidelines.
Children
Several studies have explored communication of BRCA test results to at-risk children. Across all studies, the rate of disclosure to children ranging in age from 4 to 25 years is approximately 50%. In general, age of offspring was the most important factor in deciding whether to disclose test results. In one study of 31 mothers disclosing their BRCA test results, 50% of the children who were informed of the results were aged 20 to 29 years and slightly more than 25% of the children were aged 19 years or younger. Sons and daughters were notified in equal numbers. Similarly, in another study of 42 female carriers of BRCA pathogenic variants, 83% of offspring older than age 18 years were told of the results, while only 21% of offspring aged 13 years or younger were told.
Several studies have also looked at the timing of disclosure to children after parents receive their test results. Although the majority of children were told within a week to several months after results disclosure, some parents chose to delay disclosure. Reasons for delaying disclosure included waiting for the child to get older, allowing time for the parent to adjust to the information, and waiting until results could be shared in person (in the case of adult children living away from home).
In one study, participants who told children younger than 13 years about their carrier status had increased distress, and those who did not tell their young children experienced a slight decrease in distress. Communication with young children was found to be influenced by developmental variables such as age and style of parent/child communication.
One study looked at the reaction of children to results disclosure or the effect on the parent-child relationship of communicating the results. With regard to offspring’s understanding of the information, almost half of parents from one study reported that their child did not appear to understand the significance of a positive test result, although older children were reported to have a better understanding. This same study also showed that 48% of parents reported at least one negative reaction in their child, ranging from anxiety or concern (22%) to crying and fear (26%). It should be noted, however, that in this study children's level of understanding and reactions to the test result were measured qualitatively and based only on the parents' perception. Also, given the retrospective design of the study, there was a potential for recall bias. There were no significant differences in emotional reaction depending on age or gender of the child. Lastly, 65% of parents reported no change in their relationship with their child, while 5 parents (22%) reported a strengthening of their relationship.
Interestingly, a large multicenter study of 869 mother-daughter pairs (the daughters were aged 6 to 13 y) found that girls with a family history of breast cancer or a familial BRCA1/BRCA2 pathogenic variant compared with those without such family histories had better psychosocial adjustment by maternal report. However, based on a combination of maternal report and direct assessment of girls aged 10 to 13 years, girls with family histories of breast cancer or familial BRCA1/BRCA2 pathogenic variants experienced greater breast cancer–specific distress and a higher perceived risk of breast cancer than their peers without such family histories. Moreover, higher daughter distress was associated with higher maternal distress. A similarly designed study in older girls, aged 11 to 19 years, found that higher breast cancer–specific distress in daughters was associated with perceived risk and maternal distress. This older age group had higher self-esteem than did their peers without a family history of breast cancer.
Another study of 187 mothers undergoing BRCA1/BRCA2 testing evaluated their need for resources to prepare for a facilitated conversation about sharing their BRCA1/BRCA2 testing results with their children. Seventy-eight percent of mothers were interested in three or more resources, including literature (93%), family counseling (86%), talking to prior participants (79%), and support groups (54%).
Testing for BRCA1/BRCA2 has been almost universally limited to adults older than 18 years. The risks of testing children for adult-onset disorders (such as breast and ovarian cancer), as inferred from developmental data on children’s medical understanding and ability to provide informed consent, have been outlined in several reports. Surveys of parental interest in testing children for adult-onset hereditary cancers suggest that parents are more eager to test their children than to be tested themselves for a breast cancer gene, suggesting potential conflicts for providers. In a general population survey in the United States, 71% of parents said that it was moderately, very, or extremely likely that if they carried a breast-cancer predisposing pathogenic variant, they would test a 13-year-old daughter now to determine her breast cancer gene status. To date, no data exist on the testing of children for BRCA1/BRCA2, though some researchers believe it is necessary to test the validity of assumptions underlying the general prohibition of testing of children for breast/ovarian cancer and other adult-onset disease genes. In one study, 20 children (aged 11–17 y) of a selected group of mothers undergoing genetic testing (80% of whom previously had breast cancer and all of whom had discussed BRCA1/BRCA2 testing with their children) completed self-report questionnaires on their health beliefs and attitudes toward cancer, feelings related to cancer, and behavioral problems. Ninety percent of children thought they would want cancer risk information as adults; half worried about themselves or a family member developing cancer. There was no evidence of emotional distress or behavioral problems. Another study by this group found that 1 month after disclosure of BRCA1/BRCA2 genetic test results, 53% of 42 enrolled mothers of children aged 8 to 17 years had discussed their result with one or more of their children. Age of the child rather than pathogenic variant status of the mother influenced whether they were told, as did family health communication style.
Prenatal diagnosis and preimplantation genetic testing
The possibility of transmitting a pathogenic variant to a child may pose a concern to families affected by HBOC, perhaps to the extent that some carriers may avoid childbearing. These concerns also may prompt women to consider using prenatal testing methods to help reduce the risk of transmission. Prenatal diagnosis is an encompassing term used to refer to any medical procedure conducted to assess the presence of a genetic disorder in a fetus. Methods include amniocentesis and chorionic villous sampling (CVS). Both procedures carry some risk of miscarriage and some evidence suggests fetal defects may result from using these tests. Moreover, discovering the fetus is a carrier for a genetic defect may impose a difficult decision for couples regarding pregnancy continuation or termination. An alternative to these tests is preimplantation genetic testing (PGT), a procedure used to test fertilized embryos for genetic disorders before uterine implantation, thereby avoiding the potential dangers associated with amniocentesis and CVS and the decision to terminate a pregnancy. Using the information obtained from the genetic testing, potential parents can decide whether or not to implant. PGT can be used to detect pathogenic variants in hereditary cancer predisposing genes, including BRCA.
In the United States, a series of studies has evaluated awareness, interest (e.g., would consider using PGT), and attitudes related to PGT among members of Facing Our Risk of Cancer Empowered (FORCE), an advocacy organization focused on individuals at increased risk of HBOC. The first study was a web-based survey of 283 members, the second included 205 attendees of the 2007 annual FORCE conference, and the third was a web-based survey of 962 members. These studies have documented low levels of awareness, with 20% to 32% of study respondents reporting having heard of PGT before study participation. With respect to interest in PGT, the first study found only 13% of women would be likely to use PGT, whereas, 33% of respondents in the subsequent FORCE studies reported that they would consider using PGT. In the third FORCE-based study (n = 962), multivariable analysis revealed PGT interest was associated with the desire to have more children, having previously had any prenatal genetic test, and previous awareness of PGT. Attitudinal predictors of interest in PGT included agreement that others at risk of HBOC should be offered PGT; the belief that PGT is acceptable for individuals at risk of HBOC; the belief that PGT information should be given to individuals at risk of HBOC; and endorsement of PGT benefits of having children without genetic variants and eliminating genetic diseases. Conversely, those who indicated that PGT was “too much like playing God” and reported that they considered PGT in the context of religion, had less interest in PGT.
It is unknown whether the attitudes of FORCE members toward PGT are representative of the majority of BRCA carriers. A cross-sectional study of 1,081 BRCA carriers, 65% of whom were recruited through FORCE and the remainder by the University of Pennsylvania, revealed that a majority of carriers were in favor of offering PGT and prenatal diagnosis to carriers (59% for PGT and 55.5% for prenatal diagnosis). Of those who indicated that their families were not complete, 41% of BRCA carriers reported that their carrier status impacted their decision about future biological children. This study also revealed that 21.5% of unpartnered BRCA carriers felt more pressure to get married.
The U.K. Human Fertilization and Embryology authority has approved the use of PGT for HBOC. In a sample of 102 women with a BRCA pathogenic variant, most were supportive of PGT but only 38% of the women who had completed their families would consider it for themselves had PGT been available, and only 14% of women who were contemplating a future pregnancy would consider PGT. In a study of 77 individuals undergoing BRCA testing as part of a multicenter cohort study in Spain, 61% of respondents reported they would consider PGT. Factors associated with PGT interest were age 40 years and older and had a prior cancer diagnosis.
In France, couples who obtain authorization from a multidisciplinary prenatal diagnosis team may access PGT free of charge as a benefit of their national health care system. However, no BRCA carriers have been authorized to use PGT. In a national study of 490 unaffected carriers of BRCA pathogenic variants of childbearing age (women aged 18–49 y; men aged 18–69 y), 16% stated that BRCA test results had altered their ongoing plans for childbearing. Upon qualitative analysis of written comments provided by some respondents, the primary impact was related to accelerating the timing of pregnancy, feelings of guilt about possibly passing on the pathogenic variant to offspring, and having future children. In response to a hypothetical scenario in which PGT was readily available, 33% of participants reported that they would undergo PGT. Factors associated with this intention were having no future reproductive plans at the time of the survey, feeling pregnancy termination was an acceptable option in the context of identifying a BRCA pathogenic variant, and having fewer cases of breast and/or ovarian cancer in the family. When presented with questions about expectations about delivery of PGT or prenatal diagnosis information, 85% of respondents felt it should be provided along with BRCA test results; 45% felt that it should be provided when carriers decide to have children. Respondents stated that they would expect this information to be delivered by cancer geneticists (92%), obstetrician/gynecologists (76%), and general practitioners (48%).
A small (N = 25) qualitative study of women of reproductive age positive for a BRCA pathogenic variant who underwent genetic testing before having children evaluated how their BRCA status influenced their attitudes about reproductive genetic testing (both PGT and prenatal diagnosis) and decisions about having children. In this study, the decision to undergo BRCA testing was primarily motivated by the desire to manage one’s personal cancer risk, rather than a desire to inform future reproductive decisions. The perceived severity of HBOC influenced concerns about passing on a BRCA pathogenic variant to children and also influenced willingness to consider PGT or prenatal diagnosis. Decisions about PGT and prenatal diagnosis also varied based on the participant's personal experiences. Most did not believe that a known BRCA pathogenic variant was a reason to terminate a pregnancy. As observed in prior studies, knowledge of reproductive options varied; however, there was a tendency among participants to view PGT as more acceptable than prenatal diagnosis with regard to termination of pregnancy. Decisions regarding the pros and cons of PGT versus prenatal diagnosis with termination of pregnancy were driven primarily by personal preferences and experiences, rather than by morality judgments. For example, women were deterred from PGT based on the need to undergo in vitro fertilization and to take hormones that might increase cancer risk and based on the observed experiences of others who underwent this procedure.
One study has examined these issues among high-risk men recruited from FORCE and Craigslist (a bulletin board website) (N = 228). Similar to the previous studies of women, only 20% of men were aware of PGT before survey participation. In a multivariate analysis, those who selected the “other” option for possible benefits of PGT compared with those who selected from several predetermined options (e.g., having children without genetic variants) and those who considered PGT in the context of religion (as opposed to health and safety) were less likely to report that they would ever consider using PGT.
Cultural/Community Effects
The recognition that BRCA1/BRCA2 pathogenic variants are prevalent, not only in breast/ovarian cancer families but also in some ethnic groups, has led to considerable discussion of the ethical, psychological, and other implications of having one’s ethnicity be a factor in determination of disease predisposition. Concerns that people will think everything is solely determined by genetic factors and the creation of a genetic underclass have been voiced. Questions about the impact on the group of being singled out as having genetic vulnerability to breast cancer have been raised. There is also confusion about who gives or withholds permission for the group to be involved in studies of their genetic identity. These issues challenge traditional views on informed consent as a function of individual autonomy.
A growing literature on the unique factors influencing a variety of cultural subgroups suggests the importance of developing culturally specific genetic counseling and educational approaches. The inclusion of members within the community of interest (e.g., breast cancer survivors, advocates, and community leaders) may enhance the development of culturally tailored genetic counseling materials. One study showed that participation in any genetic counseling (culturally mediated or standard approaches) reduced perceived risk of developing breast cancer.
Ethical Concerns
The human implications of the ethical issues raised by the advent of genetic testing for breast/ovarian cancer susceptibility are described in case studies, essays, and research reports. Issues about rights and responsibilities in families concerning the spread of information about genetic risk promise to be major ethical and legal dilemmas in the coming decades.
Studies have shown that 62% of studied family members were aware of the family history and that 88% of hereditary breast/ovarian cancer family members surveyed have significant concerns about privacy and confidentiality. Concern about cancer in third-degree relatives, or relatives further removed, did not differ from concern about cancer in the proband's FDRs or SDRs. Only half of surveyed FDRs of women with breast or ovarian cancer felt that written permission should be required to disclose BRCA1/BRCA2 test results to a spouse or immediate family member. Attitudes toward testing varied by ethnicity, previous exposure to genetic information, age, optimism, and information style. Altruism is a factor motivating genetic testing in some people. Many professional groups have made recommendations regarding informed consent. There is some evidence that not all practitioners are aware of or follow these guidelines. Research shows that many BRCA1/BRCA2 genetic testing consent forms do not fulfill recommendations by professional groups about the 11 areas that should be addressed, and they omit highly relevant points of information. In a study of women with a history of breast or ovarian cancer, the interviews yielded that the women reported feeling inadequately prepared for the ethical dilemmas they encountered when imparting genetic information to family members. These data suggest that more preparation about disclosure to family members before testing reduces the emotional burden of disseminating genetic information to family members. Patients and health care providers would benefit from enhanced consideration of the ethical issues of warning family members about hereditary cancer risk. (Refer to the PDQ summaries Cancer Genetics Risk Assessment and Counseling and Cancer Genetics Overview for more information about the ethics of cancer genetics and genetic testing.)
Psychosocial Aspects of Cancer Risk Management for Hereditary Breast and Gynecologic Cancers
Decision aids for individuals considering risk management options for hereditary breast and gynecologic cancers
A systematic review of decision aids for BRCA1/BRCA2 carriers suggested that these tools are beneficial during the decision-making process. BRCA1/BRCA2 carriers who used decision aids had lower decisional conflict (regarding treatment decisions), were more likely to reach risk-management decisions, and were more satisfied with their decisions than those who did not use decision aids. There were mixed or null findings regarding information-related outcomes like participant knowledge and whether there were consistent differences in actual risk-management choices.
A telephone-based intervention study used integrated motivational interviewing techniques and was led by genetic counselors. This study found no differences in rates of RRSO among BRCA1/BRCA2 carriers in the intervention arm and the control arm after 1 year. However, women in the intervention arm had significantly lower decisional conflict and higher knowledge scores after 1 year. Two years after the intervention, 53.9% of women in the intervention arm had undergone RRSO, while 32.6% of women in the control arm had undergone RRSO.
Uptake of cancer risk management options
Several studies have examined uptake and adherence to cancer risk management options in individuals who have had genetic counseling and testing for BRCA1 and BRCA2 pathogenic variants. Outcomes vary across studies and include the following: 1) uptake/adherence to screening options (mammography, magnetic resonance imaging [MRI]) and 2) selection of risk-reducing options (i.e., risk-reducing mastectomy [RRM] and/or RRSO).
Most studies reported outcomes by pathogenic variant carrier status. Follow-up time after participants received their genetic test results also varied across studies, ranging from 12 months to several years. The Hereditary Breast Cancer Clinical Study Group assessed uptake of cancer risk management options in 6,223 women with a BRCA1/BRCA2 pathogenic variant. These women were 25 to 80 years old and lived in 10 different countries (including the United States). Data from this study revealed the following:
Mammography
- In a personal communication, the first author shared that mammography uptake was defined as participants' self-reporting of mammography screenings after they received their genetic test results (K. Metcalfe, PhD, email, May 30, 2023). Mammography uptake was 82%, but this value diminished over time. The authors suggested that this decrease in mammography uptake may have been due to an increase in breast MRI uptake in participating countries, like Poland, where use of MRI exceeds the use of mammography.
MRI
- In a personal communication, the first author shared that MRI uptake was defined as participants' self-reporting of screening MRIs after they received their genetic test results (K. Metcalfe, PhD, email, May 30, 2023). The rate of screening MRI increased in women tested from Canada, Poland, and the United States from 2009 and onward (81.3%) when compared with women who tested prior to 2009 (69.5%). Overall, 72.8% of women in this study reported receiving a breast MRI in the past year.
Risk-reducing mastectomy (RRM)
- Uptake of RRM in women without a history of breast cancer was 27.8%, with RRM occurring at a mean age of 41.8 years (range, 19–78 y).
- The rate of RRM uptake was the highest in the United States (49.9%).
Risk-reducing bilateral salpingo-oophorectomy (RRSO)
- Uptake of RRSO varied slightly by gene, with 62.8% of BRCA1 carriers and 69.7% of BRCA2 carriers receiving RRSO.
- Uptake of RRSO was 70.7% in women with personal histories of breast cancer.
- For women who chose RRSO, the age of surgery also varied by gene. In BRCA1 carriers, 7.2% had RRSO performed before age 35 years, whereas in BRCA2 carriers, 37.8% had RRSO performed before age 45 years.
Chemoprevention
- Use of tamoxifen or raloxifene for risk reduction in women without histories of breast cancer was 6.3% (n = 155/2463).
- Chemoprevention use was highest in the United States (15%), with use occurring in 12.7% of BRCA1 carriers and in 17.4% of BRCA2 carriers.
In summary, breast cancer screening with mammogram in BRCA1/BRCA2 carriers may be suboptimal and can diminish over time. A portion of this decline is associated with an increase in breast MRI use, and a corresponding decrease in mammography screening, in certain countries. However, the uptake of breast MRI in BRCA1/BRCA2 carriers was also suboptimal.
Women were more likely to undergo RRM when testing from 2009 and onward (30.3%) when compared with those who tested prior to 2009 (26.9%; P = .04). Decisions about RRM may be influenced by personal factors, such as young patient age. When compared with women aged 30 to 39 years, women aged 50 to 59 years had lower rates of RRM (statistically significant) at 2, 5, and 10 years post–genetic testing. Other factors that may influence decisions about RRM include the following: a family history of breast cancer, psychosocial factors (such as a desire to reduce cancer-related distress), recommendation from a health care provider, cultural/health care system–related factors, intact ovaries, and a recent diagnosis of breast/ovarian cancer. An individual’s choice to have RRM also appears to be influenced by pretreatment genetic education and counseling, regardless of the patient's genetic test results. Additionally, when those diagnosed with breast cancer had BRCA1/BRCA2 genetic test results at the time of surgical decision-making, this resulted in increased uptake of bilateral mastectomy (OR, 8.23; 95% CI, 2.55–26.59; P< .001).
Uptake of RRSO was associated with increasing age, White race, having children, a family history of ovarian cancer, a personal history of mastectomy, higher perceived risk of ovarian cancer, and higher perceived benefit of RRSO. The decision to have RRSO also varied based on pathogenic variant carrier status, with carriers of BRCA1 pathogenic variants (N = 226) undergoing RRSO more frequently than those with BRCA2 pathogenic variants (N = 188; P = .01). In this study, a small proportion of women who were previously healthy developed breast cancer and chose to have RRSO after their cancer diagnoses (N = 14/60; 23.3%).
BRCA testing, when offered to women newly diagnosed with breast cancer, has been shown to influence surgical decision making in that carriers are more likely to opt for bilateral mastectomy compared with noncarriers. Predictors of contralateral RRM before genetic counseling and testing included younger age at breast cancer diagnosis, more time since diagnosis, having at least one affected FDR, and not being employed full-time. In the year after disclosure of test results, 18% of women who tested positive for a BRCA1/BRCA2 pathogenic variant and 2% of those whose test results were uninformative underwent contralateral RRM. Predictors of contralateral RRM after genetic testing included younger age at breast cancer diagnosis, higher cancer-specific distress before genetic counseling, and having a positive BRCA1/BRCA2 test result. In this study, contralateral RRM was not associated with distress at 1 year after disclosure of genetic test results. A retrospective chart review evaluated uptake of bilateral mastectomies in 110 women who underwent BRCA1/BRCA2 genetic testing before making surgical decisions about the treatment of newly diagnosed breast cancer. Carriers of BRCA pathogenic variants were more likely to undergo bilateral mastectomies than were women in whom no variant was detected (83% vs. 37%; P = .046). The only predictor of contralateral RRM in women without a pathogenic variant was being married (P = .03). Age, race, parity, disease stage and biomarkers, increased mammographic breast density, and breast MRI did not influence contralateral RRM decisions at the time of primary surgical treatment.
A study conducted from 2006 to 2014 in 11 U.S. academic and community centers of 897 women, aged 40 years and younger at breast cancer diagnosis, found that rates of BRCA genetic testing have increased over time. Within 1 year after diagnosis, 87% of the sample underwent BRCA testing. The rate increased from 77% of newly diagnosed women tested in 2006 to 95% of women tested in 2013. Among women who tested positive for a pathogenic BRCA variant and stated that testing affected their surgery decisions (n = 88), 86% underwent bilateral mastectomy compared with 51% of noncarriers (P< .001). Among untested women, about one-third reported that they were not told by a health care provider that they were candidates for BRCA testing; yet, according to national guidelines, all were eligible for testing solely on the basis of their age at diagnosis.
A systematic review of risk-reducing salpingectomy with delayed oophorectomy (RRDSO) found that the avoidance of surgical menopause, preservation of fertility, concerns about sexual functioning, a family history of breast cancer, and an interest to avoid HRT were all related to being more likely to accept this option. Those concerned with surgical timing, complications, and ongoing cancer risk were less likely to opt for RRDSO. In a small cohort of premenopausal BRCA1/BRCA2 carriers (n = 43), 44% chose to pursue RRDSO, 28% chose to pursue RRSO, and 28% chose to pursue only screening. One year after surgery, cancer worry had decreased among all of those who elected to have surgery. Surgical complications were not reported.
Cancer screening and risk-reducing behaviors
Data are now emerging regarding uptake and adherence to cancer risk management recommendations such as screening and risk-reducing interventions. Cancer screening adherence and risk-reduction behaviors as defined by the National Comprehensive Cancer Network Guidelines were assessed in a cross-sectional study of 214 women with a personal history (n = 134) or family history (n = 80) of breast or ovarian cancer. Among unaffected women older than 40 years, 10% had not had a mammogram or clinical breast examination (CBE) in the previous year and 46% did not practice breast self-examination (BSE). Among women previously affected with breast or ovarian cancer, 21% had not had a mammogram, 32% had not had a CBE, and 39% did not practice BSE.
Three hundred and twelve women who were counseled and tested for BRCA pathogenic variants between 1997 and 2005 responded to a survey regarding their perception of genetic testing for HBOC. The survey included questions on risk reduction options, including screening and risk-reducing surgeries. Two hundred and seventeen (70%) of the women had been diagnosed with breast cancer, and 86 (28%) tested positive for a pathogenic variant in either the BRCA1 or BRCA2 gene. None of the BRCA-positive women agreed that mammograms are difficult procedures because of the discomfort, while 11 (5.4%) of the BRCA-negative women did agree with this statement. Both groups (BRCA-positive and BRCA-negative) agreed that risk-reducing surgeries provide the best means for lowering cancer risk and worry, and most patients in both groups expressed the belief that RRM is not too drastic, too scary, or too disfiguring.
A prospective study from the United Kingdom examined the psychological impact of mammographic screening in 1,286 women aged 35 to 49 years who have a family history of breast cancer and were participants in a multicenter screening program. Mammographic abnormalities that required additional evaluation were detected in 112 women. These women, however, did not show a statistically significant increase in cancer worry or negative psychological consequences as a result of these findings. The 1,174 women who had no mammographic abnormality detected experienced a decrease in cancer worry and a decrease in negative psychological consequences compared with baseline after receipt of their results. At 6 months, the entire cohort had experienced a decrease in measures of cancer worry and psychological consequences of breast screening.
A qualitative study explored health care professionals’ views regarding the provision of information about health protective behaviors (e.g., exercise and diet). Seven medical specialists and ten genetic counselors were interviewed during a focus group or individually. The study reported wide variation in the content and extent of information provided about health-protective behaviors and in general, participants did not consider it their role to promote such behaviors in the context of a genetic counseling session. There was agreement, however, about the need to form consensus about provision of such information both within and across risk assessment clinics.
Not all studies specify whether screening uptake rates fall within recommended guidelines for the targeted population or the specific clinical scenario, nor do they report on other variables that may influence cancer screening recommendations. For example, women who have a history of atypical ductal hyperplasia would be advised to follow screening recommendations that may differ from those of the general population.
Psychosocial Outcome Studies
Psychosocial outcomes associated with risk-reducing mastectomy (RRM)
A prospective study conducted in the Netherlands found that among 26 carriers of BRCA1/BRCA2 pathogenic variants, the 14 women who chose mastectomy had higher distress both before test result disclosure and 6 and 12 months later, compared with the 12 carriers who chose surveillance and compared with 53 women negative for a pathogenic variant. Overall, however, anxiety declined in women undergoing RRM; at 1 year, their anxiety scores were closer to those of women choosing surveillance and to the scores of women negative for a pathogenic variant. Interestingly, women opting for RRM had lower pretest satisfaction with their breasts and general body image than carriers who opted for surveillance or noncarriers of BRCA1/BRCA2 pathogenic variants. Of the women who had an RRM, all but one did not regret the decision at 1 year posttest disclosure, but many had difficulties with body image, sexual interest and functioning, and self-esteem. The perception that doctors had inadequately informed them about the consequences of RRM was associated with regret. At the 5-year follow-up, women who had undergone RRM had less favorable body image and changes in sexual relationships, but also had a significant reduction in the fear of developing cancer. In a study of 78 women who underwent risk-reducing surgery (including BRCA1/BRCA2 carriers and women who were from high-risk families with no detectable BRCA1/BRCA2 pathogenic variant), cancer-specific and general distress were assessed 2 weeks before surgery and at 6 and 12 months postsurgery. The sample included women who had RRM and RRSO alone and women who had both surgeries. There was no observable increase in distress over the 1-year period.
Mixed psychosocial outcomes were reported in a follow-up study (mean, 14 y) of 609 women who received RRM at the Mayo Clinic. Seventy percent were satisfied with RRM, 11% were neutral, and 19% were dissatisfied. Eighteen percent believed that if they had the choice to make again, they probably or definitely would not have an RRM. About three-quarters said their worry about cancer was diminished by surgery. One-half reported no change in their satisfaction with body image; 16% reported improved body image after surgery. Thirty-six percent said they were dissatisfied with their body image after RRM. About one-quarter of the women reported adverse impact of RRM on their sexual relationships and sense of femininity, and 18% had diminished self-esteem. Factors most strongly associated with satisfaction with RRM were postsurgical satisfaction with appearance, reduced stress, no reconstruction or lack of problems with implants, and no change or improvement in sexual relationships. Women who cited physician advice as the primary reason for choosing RRM tended to be dissatisfied after RRM.
A study of 60 healthy women who underwent RRM measured levels of satisfaction, body image, sexual functioning, intrusion and avoidance, and current psychological status at a mean of 4 years and 4 months postsurgery. Of this group, 76.7% had either a strong family history (21.7%) or carried a BRCA1 or BRCA2 pathogenic variant (55%). Overall, 97% of the women surveyed were either satisfied (17%) or extremely satisfied (80%) with their decision to have RRM, and all but one participant would recommend this procedure to other women. Most women (66.7%) reported that surgery had no impact on their sexual life, although 31.7% reported a worsening sexual life, and 76.6% reported either no change in body image or an improvement in body image, regardless of whether reconstruction was performed. Worsening self-image was reported by 23.3% of women after surgery. Women’s mean distress levels after surgery were only slightly above normal levels, although those women who continued to perceive their postsurgery breast cancer risk as high had higher mean levels of global and cancer-related distress than those who perceived their risk as low. Additionally, carriers of BRCA1 and BRCA2 pathogenic variants and women with a strong family history of breast and/or ovarian cancer had higher mean levels of cancer-related distress than women with a limited family history.
Very little is known about how the results of genetic testing affect treatment decisions at the time of cancer diagnosis. Two studies explored genetic counseling and BRCA1/BRCA2 genetic testing at the time of breast cancer diagnosis. One of these studies found that genetic testing at the time of diagnosis significantly altered surgical decision making, with more pathogenic variant carriers than noncarriers opting for bilateral mastectomy. Bilateral RRM was chosen by 48% of women with a known pathogenic variant and by 100% of women with a known pathogenic variant in a smaller series of women undergoing testing at the time of diagnosis. Of women in whom no pathogenic variant was found, 24% also opted for bilateral RRM. Four percent of the test decliners also underwent bilateral RRM. Among carriers of pathogenic variants, predictors of bilateral RRM included whether patients reported that their physicians had recommended BRCA1/BRCA2 testing and bilateral RRM before testing, and whether they received a positive test result. Data are lacking on quality-of-life outcomes for women who undergo RRM after genetic testing that is performed at the time of diagnosis.
A prospective study from the Netherlands evaluated long-term psychological outcomes of offering women with breast cancer genetic counseling and, if indicated, genetic testing at the onset of breast radiation for treatment of their primary breast cancer. Of those who were approached for counseling, some underwent genetic testing and chose to receive their result (n = 58), some were approached but did not fulfill referral criteria (n = 118), and some declined the option of counseling/testing (n = 44). Another subset of women undergoing radiation therapy was not approached for counseling (n = 182) but was followed using the same measures. Psychological distress was measured at baseline and at 4, 11, 27, and 43 weeks after initial consultation for radiation therapy. No differences were detected in general anxiety, depression or breast cancer–specific distress across all four groups.
A retrospective questionnaire study of 583 women with a personal and family history of breast cancer and who underwent contralateral RRM between 1960 and 1993 measured overall satisfaction after mastectomy and factors influencing satisfaction and dissatisfaction with this procedure. The mean time of follow-up was 10.3 years after risk-reducing surgery. Overall, 83% of all participants stated they were satisfied or very satisfied, 8% were neutral, and 9% were dissatisfied with contralateral RRM. Most women also reported favorable effects or no change in their self-esteem, level of stress, and emotional stability after surgery (88%, 83%, and 88%, respectively). Despite the high levels of overall satisfaction, 33% reported negative body image, 26% reported a reduced sense of femininity, and 23% reported a negative effect on sexual relationships. The type of surgical procedure also affected levels of satisfaction. The authors attributed this difference to the high rate of unanticipated reoperations in the group of women having subcutaneous mastectomy (43%) versus the group having simple mastectomy (15%) (P< .0001). Limitations to this study are mostly related to the time period during which participants had their surgery (i.e., availability of surgical reconstructive option). None of these women had genetic testing for pathogenic variants in the BRCA1/BRCA2 genes. Nevertheless, this study shows that while most women in this group were satisfied with contralateral RRM, all women reported at least one adverse outcome.
A retrospective survey of 137 BRCA carriers examined the psychosocial impact of preserving the nipple-areolar complex (NAC) in women with bilateral RRM. The study found that body image and sexual well-being differed significantly based on the type of RRM the women underwent. Women with NAC preservation were more satisfied with their breasts (72% vs. 61%), were more satisfied with the surgical outcome (85% vs. 74%) and had greater sexual well-being (68% vs. 52%) than women without NAC preservation. No differences in cancer-related distress, anxiety, depression, or risk perceptions were observed between the two groups. Oncological outcomes of nipple-sparing mastectomy in BRCA carriers have not been inferior to RRM without NAC preservation. (Refer to the Risk-reducing strategies for breast cancer section in BRCA1 and BRCA2: Cancer Risks and Management for more information.)
Another study compared long-term quality-of-life outcomes in 195 women after bilateral RRM performed between 1979 and 1999 versus 117 women at high risk of breast cancer opting for screening. No statistically significant differences were detected between the groups for psychosocial outcomes. Eighty-four percent of those opting for surgery reported satisfaction with their decision. Sixty-one percent of women from both the surgery and screening groups reported being very much or quite a bit contented with their quality of life.
In a study of psychosocial outcomes associated with RRM and immediate reconstruction, 61 high-risk women (27 carriers of pathogenic variants, others with high-risk family history), 31 of whom had a prior history of breast cancer, were evaluated on average 3 to 4 years after surgery. The study utilized questions designed to elicit yes versus no responses and found that the surgery was well-tolerated with 83% of participants reporting that the results of their reconstructive surgery were as they expected, 90% reporting that they had received adequate preoperative information, none reporting that they regretted the surgery, and all reporting that they would choose the same route if they had to do it again. Satisfaction with the results ranged from 74% satisfied with the shape of their breasts to 89% satisfied with the appearance of the scarring. Comparison of this group to normative samples on quality-of-life indicators (Short Form 36 Health Survey Questionnaire; Hospital Anxiety and Depression Scale questionnaire scores) indicated no reductions in quality of life in these women.
A qualitative study examining material on the FORCE website posted by 21 high-risk women (BRCA1/BRCA2-positive) undergoing RRM showed that these women anticipated and received negative reactions from friends and family regarding the surgery, and that they managed disclosure in ways to maintain emotional support and self-protection for their decision. Many of the women expressed a relief from intrusive breast cancer thoughts and worry, and were satisfied with the cosmetic result of their surgery.
In contrast, another study examined long-term psychosocial outcomes in 684 women who had had bilateral or contralateral RRM on average 9 years before assessment. A majority of women (59%) also had reconstructive surgery. Interestingly, based on a Likert scale, 85% of women reported that they were satisfied or very satisfied with their decision to have an RRM. However, in qualitative interviews, many women went on to describe dissatisfaction or negative psychosocial outcomes associated with surgery. The authors coded the responses as negative when women reported still being anxious about their breast cancer risk and/or reported negative feelings about their body image, pain, and sexuality. Seventy-nine percent of the women providing negative comments and 84% of those making mixed comments (mixture of satisfaction and dissatisfaction) responded that they were either satisfied or very satisfied with their decision. Twice as many women with bilateral mastectomy made negative and mixed comments than did women with contralateral mastectomy. The areas of most concern were body image, problems with breast implants, pain after surgery, and sexuality. The authors proposed that those who had undergone contralateral procedures had already been treated for cancer, while those who had undergone bilateral procedures had not been treated previously, and this may partially account for the differences in satisfaction between the two groups. These findings suggest that women's satisfaction with RRM may be tempered by their complex reactions over time.
In a qualitative study of 108 women who underwent or were considering RRM, more than half of those who had RRM felt that presurgical consultation with a psychologist was advisable; nearly two-thirds thought that postsurgical consultation was also appropriate. All of the women who were considering RRM believed that psychological consultation before surgery would facilitate decision-making.
Psychosocial outcomes associated with risk-reducing salpingo-oophorectomy (RRSO)
Prior studies on the impact of RRSO suggest some variability in outcomes based on menopausal status:
- A Canadian prospective study examined the impact of RRSO on menopausal symptoms and sexual functioning in a sample of 140 women with known BRCA1/BRCA2 pathogenic variants. This study compared outcomes before surgery and after surgery (an average of 3.5 y later). RRSO was associated with an increase in menopausal symptoms and a decrease in reported sexual functioning in premenopausal women (n = 93). However, differences in overall quality of life (QOL) were not reported in this group. HRT reduced the adverse effects associated with RRSO but did not fully eliminate them. In postmenopausal women (n = 47), RRSO resulted in an increase in physical symptoms (i.e., insomnia and fatigue) and decreases in both sexual functioning and overall QOL.
- A cross-sectional study analyzed 244 women with BRCA1/BRCA2 pathogenic variants who were aged 40 years or older. BRCA1/BRCA2 carriers who did not undergo RRSO had more cancer worry than women who had undergone RRSO, but fewer menopausal symptoms. Those who underwent RRSO and those who did not had similar levels of sexual activity and functioning. Comparing women with pre- vs. postmenopausal RRSO, premenopausal women were more likely to be sexually active, to report menopausal symptoms, and to have depression than postmenopausal women. However, premenopausal and postmenopausal women had similar levels of sexual functioning. For more information, see the Risk-reducing salpingo-oophorectomy for ovarian cancer risk reduction section in BRCA1 and BRCA2: Cancer Risks and Management.
- A cross-sectional study of 641 women (66% of whom were BRCA1/BRCA2 carriers) who underwent RRSO before or after menopause found no long-term association between premenopausal RRSO and measures of cognitive function.
Behavioral Outcomes
A study of screening behaviors of 216 self-referred, high-risk (>10% risk of carrying a BRCA1/BRCA2 pathogenic variant) women who are members of hereditary breast cancer families found a range of screening practices. Even the presence of known pathogenic variants in their families was not associated with good adherence to recommended screening practices. Sixty-nine percent of women aged 50 to 64 years and 83% of women aged 40 to 49 years had had a screening mammogram in the previous year. Twenty percent of participants had ever had a CA-125 test and 31% had ever had a pelvic ultrasound or TVUS. Further analysis of this study population looking specifically at 107 women with informative BRCA test results found good use of breast cancer screening, though the uptake rate in younger carriers is lower. The reason for the lower uptake rate was not explored in this study. One survey of screening behaviors among women at increased risk of breast/ovarian cancer identified physician recommendations as a significant factor in adherence to screening.
While motivations cited for pursuing genetic testing often include the expectation that carriers of pathogenic variants will be more compliant with breast and/or ovarian screening recommendations, limited data exist about whether participants in genetic testing alter their screening behaviors over time and about other variables that may influence those behaviors, such as insurance coverage and physician recommendations or attitudes. The impact of cancer genetic counseling on screening behaviors was assessed in a U.K. study of 293 women followed for 12 months postcounseling at four cancer genetics clinics. BSE, CBE, and mammography were significantly increased after counseling; however, gaps in adherence to recommendations were noted: 38% of women aged 35 to 49 years had not had a mammogram by 12 months postcounseling. BSE was not done by most women at the recommended time and frequency.
This is a critical issue not only for women testing positive, but also for adherence to screening for those testing negative and those who have received indeterminate results or choose not to receive their results. It is possible that adherence actually diminishes with a decrease in the perceived risk that may result from a negative genetic test result.
In addition, while there is still some question regarding the link between cancer-related worry and breast cancer screening behavior, accumulating evidence appears to support a linear rather than a curvilinear relationship. That is, for some time, the data were not consistent; some data supported the hypothesis that mild-to-moderate worry may increase adherence, while excessive worry may actually decrease the utilization of recommended screening practices. Other reports support the notion that a linear relationship is more likely; that is, more worry increases adherence to screening recommendations. Few studies, however, have followed women to assess their health behaviors after genetic testing. Thus, a negative test result leading to decreased worry could theoretically result in decreased screening adherence. A large study found that patient compliance with screening practices was not related to general or screening-specific anxiety—with the exception of BSE, for which compliance is negatively associated with procedure-specific anxiety. Further research designed to clarify this potential concern would highlight the need for comprehensive genetic counseling to discuss the need for follow-up screening.
Further complicating this area of research are issues such as the baseline rate of mammography adherence among women older than 40 or 50 years before genetic testing. More specifically, the ability to note a significant difference in adherence on this measure may be affected by the high adherence rate to this screening behavior before genetic testing by women undergoing such testing. It may be easier to find significant changes in mammography use among women with a family history of breast cancer who test positive. Finally, adherence over time will likely be affected by how women undergoing genetic testing and their caregivers perceive the efficacy of many of the screening options in question, such as mammography for younger women, BSE, and ovarian cancer screening (periodic vaginal ultrasound and serum CA-125 measurements), along with the value of preventive interventions.
The issue of screening decision-making and adherence among women undergoing genetic testing for breast and ovarian cancer is the subject of several ongoing trials, and an area of much needed ongoing study.
References
- Ropka ME, Wenzel J, Phillips EK, et al.: Uptake rates for breast cancer genetic testing: a systematic review. Cancer Epidemiol Biomarkers Prev 15 (5): 840-55, 2006.
- Schwartz MD, Lerman C, Brogan B, et al.: Utilization of BRCA1/BRCA2 mutation testing in newly diagnosed breast cancer patients. Cancer Epidemiol Biomarkers Prev 14 (4): 1003-7, 2005.
- Kieran S, Loescher LJ, Lim KH: The role of financial factors in acceptance of clinical BRCA genetic testing. Genet Test 11 (1): 101-10, 2007.
- Susswein LR, Skrzynia C, Lange LA, et al.: Increased uptake of BRCA1/2 genetic testing among African American women with a recent diagnosis of breast cancer. J Clin Oncol 26 (1): 32-6, 2008.
- Olaya W, Esquivel P, Wong JH, et al.: Disparities in BRCA testing: when insurance coverage is not a barrier. Am J Surg 198 (4): 562-5, 2009.
- Levy DE, Byfield SD, Comstock CB, et al.: Underutilization of BRCA1/2 testing to guide breast cancer treatment: black and Hispanic women particularly at risk. Genet Med 13 (4): 349-55, 2011.
- Landsbergen K, Verhaak C, Kraaimaat F, et al.: Genetic uptake in BRCA-mutation families is related to emotional and behavioral communication characteristics of index patients. Fam Cancer 4 (2): 115-9, 2005.
- Denayer L, Boogaerts A, Philippe K, et al.: BRCA1/2 predictive testing and gender: uptake, motivation and psychological characteristics. Genet Couns 20 (4): 293-305, 2009.
- Meijers-Heijboer EJ, Verhoog LC, Brekelmans CT, et al.: Presymptomatic DNA testing and prophylactic surgery in families with a BRCA1 or BRCA2 mutation. Lancet 355 (9220): 2015-20, 2000.
- Wakefield CE, Ratnayake P, Meiser B, et al.: "For all my family's sake, I should go and find out": an Australian report on genetic counseling and testing uptake in individuals at high risk of breast and/or ovarian cancer. Genet Test Mol Biomarkers 15 (6): 379-85, 2011.
- Schlich-Bakker KJ, ten Kroode HF, Wárlám-Rodenhuis CC, et al.: Barriers to participating in genetic counseling and BRCA testing during primary treatment for breast cancer. Genet Med 9 (11): 766-77, 2007.
- Vadaparampil ST, McIntyre J, Quinn GP: Awareness, perceptions, and provider recommendation related to genetic testing for hereditary breast cancer risk among at-risk Hispanic women: similarities and variations by sub-ethnicity. J Genet Couns 19 (6): 618-29, 2010.
- Meiser B: Psychological impact of genetic testing for cancer susceptibility: an update of the literature. Psychooncology 14 (12): 1060-74, 2005.
- Pasacreta JV: Psychosocial issues associated with genetic testing for breast and ovarian cancer risk: an integrative review. Cancer Invest 21 (4): 588-623, 2003.
- Simon MS, Petrucelli N: Hereditary breast and ovarian cancer syndrome : the impact of race on uptake of genetic counseling and testing. Methods Mol Biol 471: 487-500, 2009.
- Hann KEJ, Freeman M, Fraser L, et al.: Awareness, knowledge, perceptions, and attitudes towards genetic testing for cancer risk among ethnic minority groups: a systematic review. BMC Public Health 17 (1): 503, 2017.
- Meiser B, Gaff C, Julian-Reynier C, et al.: International perspectives on genetic counseling and testing for breast cancer risk. Breast Dis 27: 109-25, 2006-2007.
- Armstrong K, Stopfer J, Calzone K, et al.: What does my doctor think? Preferences for knowing the doctor's opinion among women considering clinical testing for BRCA1/2 mutations. Genet Test 6 (2): 115-8, 2002 Summer.
- McCuaig JM, Greenwood CM, Shuman C, et al.: Breast and ovarian cancer: the forgotten paternal contribution. J Genet Couns 20 (5): 442-9, 2011.
- Burke W, Culver J, Pinsky L, et al.: Genetic assessment of breast cancer risk in primary care practice. Am J Med Genet A 149A (3): 349-56, 2009.
- Mouchawar J, Klein CE, Mullineaux L: Colorado family physicians' knowledge of hereditary breast cancer and related practice. J Cancer Educ 16 (1): 33-7, 2001.
- Yong MC, Zhou XJ, Lee SC: The importance of paternal family history in hereditary breast cancer is underappreciated by health care professionals. Oncology 64 (3): 220-6, 2003.
- Bellcross CA, Leadbetter S, Alford SH, et al.: Prevalence and healthcare actions of women in a large health system with a family history meeting the 2005 USPSTF recommendation for BRCA genetic counseling referral. Cancer Epidemiol Biomarkers Prev 22 (4): 728-35, 2013.
- Weitzel JN, McCaffrey SM, Nedelcu R, et al.: Effect of genetic cancer risk assessment on surgical decisions at breast cancer diagnosis. Arch Surg 138 (12): 1323-8; discussion 1329, 2003.
- Kurian AW, Griffith KA, Hamilton AS, et al.: Genetic Testing and Counseling Among Patients With Newly Diagnosed Breast Cancer. JAMA 317 (5): 531-534, 2017.
- Childers CP, Childers KK, Maggard-Gibbons M, et al.: National Estimates of Genetic Testing in Women With a History of Breast or Ovarian Cancer. J Clin Oncol 35 (34): 3800-3806, 2017.
- Pal T, Bonner D, Kim J, et al.: Early onset breast cancer in a registry-based sample of African-american women: BRCA mutation prevalence, and other personal and system-level clinical characteristics. Breast J 19 (2): 189-92, 2013 Mar-Apr.
- Weldon CB, Trosman JR, Gradishar WJ, et al.: Barriers to the use of personalized medicine in breast cancer. J Oncol Pract 8 (4): e24-31, 2012.
- Hafertepen L, Pastorino A, Morman N, et al.: Barriers to genetic testing in newly diagnosed breast cancer patients: Do surgeons limit testing? Am J Surg 214 (1): 105-110, 2017.
- Katz SJ, Ward KC, Hamilton AS, et al.: Gaps in Receipt of Clinically Indicated Genetic Counseling After Diagnosis of Breast Cancer. J Clin Oncol 36 (12): 1218-1224, 2018.
- Schwartz MD, Peshkin BN, Isaacs C, et al.: Randomized trial of proactive rapid genetic counseling versus usual care for newly diagnosed breast cancer patients. Breast Cancer Res Treat 170 (3): 517-524, 2018.
- Lerman C, Hughes C, Benkendorf JL, et al.: Racial differences in testing motivation and psychological distress following pretest education for BRCA1 gene testing. Cancer Epidemiol Biomarkers Prev 8 (4 Pt 2): 361-7, 1999.
- Armstrong K, Micco E, Carney A, et al.: Racial differences in the use of BRCA1/2 testing among women with a family history of breast or ovarian cancer. JAMA 293 (14): 1729-36, 2005.
- Kinney AY, Simonsen SE, Baty BJ, et al.: Acceptance of genetic testing for hereditary breast ovarian cancer among study enrollees from an African American kindred. Am J Med Genet A 140 (8): 813-26, 2006.
- Halbert CH, Kessler L, Stopfer JE, et al.: Low rates of acceptance of BRCA1 and BRCA2 test results among African American women at increased risk for hereditary breast-ovarian cancer. Genet Med 8 (9): 576-82, 2006.
- Thompson HS, Valdimarsdottir HB, Duteau-Buck C, et al.: Psychosocial predictors of BRCA counseling and testing decisions among urban African-American women. Cancer Epidemiol Biomarkers Prev 11 (12): 1579-85, 2002.
- Kinney AY, Gammon A, Coxworth J, et al.: Exploring attitudes, beliefs, and communication preferences of Latino community members regarding BRCA1/2 mutation testing and preventive strategies. Genet Med 12 (2): 105-15, 2010.
- Sussner KM, Edwards T, Villagra C, et al.: BRCA genetic counseling among at-risk Latinas in New York City: new beliefs shape new generation. J Genet Couns 24 (1): 134-48, 2015.
- Zimmerman RK, Tabbarah M, Nowalk MP, et al.: Racial differences in beliefs about genetic screening among patients at inner-city neighborhood health centers. J Natl Med Assoc 98 (3): 370-7, 2006.
- MacNew HG, Rudolph R, Brower ST, et al.: Assessing the knowledge and attitudes regarding genetic testing for breast cancer risk in our region of southeastern Georgia. Breast J 16 (2): 189-92, 2010.
- Sussner KM, Thompson HS, Jandorf L, et al.: The influence of acculturation and breast cancer-specific distress on perceived barriers to genetic testing for breast cancer among women of African descent. Psychooncology 18 (9): 945-55, 2009.
- Edwards TA, Thompson HS, Kwate NO, et al.: Association between temporal orientation and attitudes about BRCA1/2 testing among women of African descent with family histories of breast cancer. Patient Educ Couns 72 (2): 276-82, 2008.
- McCarthy AM, Bristol M, Domchek SM, et al.: Health Care Segregation, Physician Recommendation, and Racial Disparities in BRCA1/2 Testing Among Women With Breast Cancer. J Clin Oncol 34 (22): 2610-8, 2016.
- Hurtado-de-Mendoza A, Jackson MC, Anderson L, et al.: The Role of Knowledge on Genetic Counseling and Testing in Black Cancer Survivors at Increased Risk of Carrying a BRCA1/2 Mutation. J Genet Couns 26 (1): 113-121, 2017.
- Cragun D, Weidner A, Lewis C, et al.: Racial disparities in BRCA testing and cancer risk management across a population-based sample of young breast cancer survivors. Cancer 123 (13): 2497-2505, 2017.
- Cragun D, Weidner A, Kechik J, et al.: Genetic Testing Across Young Hispanic and Non-Hispanic White Breast Cancer Survivors: Facilitators, Barriers, and Awareness of the Genetic Information Nondiscrimination Act. Genet Test Mol Biomarkers 23 (2): 75-83, 2019.
- Lerman C, Hughes C, Lemon SJ, et al.: What you don't know can hurt you: adverse psychologic effects in members of BRCA1-linked and BRCA2-linked families who decline genetic testing. J Clin Oncol 16 (5): 1650-4, 1998.
- Lodder L, Frets PG, Trijsburg RW, et al.: Attitudes and distress levels in women at risk to carry a BRCA1/BRCA2 gene mutation who decline genetic testing. Am J Med Genet 119A (3): 266-72, 2003.
- Foster C, Evans DG, Eeles R, et al.: Non-uptake of predictive genetic testing for BRCA1/2 among relatives of known carriers: attributes, cancer worry, and barriers to testing in a multicenter clinical cohort. Genet Test 8 (1): 23-9, 2004.
- McInerney-Leo A, Biesecker BB, Hadley DW, et al.: BRCA1/2 testing in hereditary breast and ovarian cancer families II: impact on relationships. Am J Med Genet A 133 (2): 165-9, 2005.
- Patenaude AF: Cancer susceptibility testing: risks, benefits, and personal beliefs. In: Clarke A, ed.: The Genetic Testing of Children. BIOS Scientific, 1998, pp 145-156.
- Richards M: The genetic testing of children: adult attitude's and children's understanding. In: Clarke A, ed.: The Genetic Testing of Children. BIOS Scientific, 1998, pp 169-179.
- Wertz DC, Fanos JH, Reilly PR: Genetic testing for children and adolescents. Who decides? JAMA 272 (11): 875-81, 1994.
- Borry P, Stultiëns L, Nys H, et al.: Attitudes towards predictive genetic testing in minors for familial breast cancer: a systematic review. Crit Rev Oncol Hematol 64 (3): 173-81, 2007.
- Hamann HA, Croyle RT, Venne VL, et al.: Attitudes toward the genetic testing of children among adults in a Utah-based kindred tested for a BRCA1 mutation. Am J Med Genet 92 (1): 25-32, 2000.
- Bradbury AR, Patrick-Miller L, Pawlowski K, et al.: Should genetic testing for BRCA1/2 be permitted for minors? Opinions of BRCA mutation carriers and their adult offspring. Am J Med Genet C Semin Med Genet 148C (1): 70-7, 2008.
- Points to consider: ethical, legal, and psychosocial implications of genetic testing in children and adolescents. American Society of Human Genetics Board of Directors, American College of Medical Genetics Board of Directors. Am J Hum Genet 57 (5): 1233-41, 1995.
- Michie S, Marteau TM: Predictive genetic testing in children: the need for psychological research. In: Clarke A, ed.: The Genetic Testing of Children. BIOS Scientific, 1998, pp 169-182.
- MacDonald DJ, Lessick M: Hereditary cancers in children and ethical and psychosocial implications. J Pediatr Nurs 15 (4): 217-25, 2000.
- Tercyak KP, Peshkin BN, Streisand R, et al.: Psychological issues among children of hereditary breast cancer gene (BRCA1/2) testing participants. Psychooncology 10 (4): 336-46, 2001 Jul-Aug.
- Winer E, Winer N, Bluman L, et al.: Attitudes and risk perceptions of women with breast cancer considering testing for BRCA1/2. [Abstract] Proceedings of the American Society of Clinical Oncology 16: A1937, 537a, 1997.
- Braithwaite D, Emery J, Walter F, et al.: Psychological impact of genetic counseling for familial cancer: a systematic review and meta-analysis. J Natl Cancer Inst 96 (2): 122-33, 2004.
- Mikkelsen EM, Sunde L, Johansen C, et al.: Psychosocial conditions of women awaiting genetic counseling: a population-based study. J Genet Couns 17 (3): 242-51, 2008.
- Dorval M, Bouchard K, Maunsell E, et al.: Health behaviors and psychological distress in women initiating BRCA1/2 genetic testing: comparison with control population. J Genet Couns 17 (4): 314-26, 2008.
- Wang C, Gonzalez R, Janz N, et al.: The role of cognitive appraisal and worry in BRCA1/2 testing decisions among a clinic population. Psychol Health 22 (6): 719-36, 2007.
- Hallowell N, Statham H, Murton F: Women's understanding of their risk of developing breast/ovarian cancer before and after genetic counseling. J Genet Couns 7 (4): 345-64, 1998.
- MacDonald DJ, Choi J, Ferrell B, et al.: Concerns of women presenting to a comprehensive cancer centre for genetic cancer risk assessment. J Med Genet 39 (7): 526-30, 2002.
- Matloff ET, Moyer A, Shannon KM, et al.: Healthy women with a family history of breast cancer: impact of a tailored genetic counseling intervention on risk perception, knowledge, and menopausal therapy decision making. J Womens Health (Larchmt) 15 (7): 843-56, 2006.
- Meiser B, Price MA, Butow PN, et al.: Misperceptions of ovarian cancer risk in women at increased risk for hereditary ovarian cancer. Fam Cancer 13 (2): 153-62, 2014.
- Bluman LG, Rimer BK, Berry DA, et al.: Attitudes, knowledge, and risk perceptions of women with breast and/or ovarian cancer considering testing for BRCA1 and BRCA2. J Clin Oncol 17 (3): 1040-6, 1999.
- Iglehart JD, Miron A, Rimer BK, et al.: Overestimation of hereditary breast cancer risk. Ann Surg 228 (3): 375-84, 1998.
- Bluman LG, Rimer BK, Regan Sterba K, et al.: Attitudes, knowledge, risk perceptions and decision-making among women with breast and/or ovarian cancer considering testing for BRCA1 and BRCA2 and their spouses. Psychooncology 12 (5): 410-27, 2003 Jul-Aug.
- McCaul KD, O'Donnell SM: Naive beliefs about breast cancer risk. Womens Health 4 (1): 93-101, 1998 Spring.
- Huiart L, Eisinger F, Stoppa-Lyonnet D, et al.: Effects of genetic consultation on perception of a family risk of breast/ovarian cancer and determinants of inaccurate perception after the consultation. J Clin Epidemiol 55 (7): 665-75, 2002.
- Davis S, Stewart S, Bloom J: Increasing the accuracy of perceived breast cancer risk: results from a randomized trial with Cancer Information Service callers. Prev Med 39 (1): 64-73, 2004.
- Katapodi MC, Dodd MJ, Lee KA, et al.: Underestimation of breast cancer risk: influence on screening behavior. Oncol Nurs Forum 36 (3): 306-14, 2009.
- Lindberg NM, Wellisch D: Anxiety and compliance among women at high risk for breast cancer. Ann Behav Med 23 (4): 298-303, 2001 Fall.
- Ritvo P, Irvine J, Robinson G, et al.: Psychological adjustment to familial-genetic risk assessment for ovarian cancer: predictors of nonadherence to surveillance recommendations. Gynecol Oncol 84 (1): 72-80, 2002.
- Meiser B, Halliday JL: What is the impact of genetic counselling in women at increased risk of developing hereditary breast cancer? A meta-analytic review. Soc Sci Med 54 (10): 1463-70, 2002.
- Green J, Richards M, Murton F, et al.: Family communication and genetic counseling: the case of hereditary breast and ovarian cancer. J Genet Couns 6 (1): 45-60, 1997.
- Quillin JM, Ramakrishnan V, Borzelleca J, et al.: Paternal relatives and family history of breast cancer. Am J Prev Med 31 (3): 265-8, 2006.
- O'Neill SM, Rubinstein WS, Wang C, et al.: Familial risk for common diseases in primary care: the Family Healthware Impact Trial. Am J Prev Med 36 (6): 506-14, 2009.
- Rubinstein WS, O'neill SM, Rothrock N, et al.: Components of family history associated with women's disease perceptions for cancer: a report from the Family Healthware™ Impact Trial. Genet Med 13 (1): 52-62, 2011.
- Theis B, Boyd N, Lockwood G, et al.: Accuracy of family cancer history in breast cancer patients. Eur J Cancer Prev 3 (4): 321-7, 1994.
- Breuer B, Kash KM, Rosenthal G, et al.: Reporting bilaterality status in first-degree relatives with breast cancer: a validity study. Genet Epidemiol 10 (4): 245-56, 1993.
- Parent ME, Ghadirian P, Lacroix A, et al.: The reliability of recollections of family history: implications for the medical provider. J Cancer Educ 12 (2): 114-20, 1997 Summer.
- Kelly KM, Shedlosky-Shoemaker R, Porter K, et al.: Cancer family history reporting: impact of method and psychosocial factors. J Genet Couns 16 (3): 373-82, 2007.
- Kerber RA, Slattery ML: Comparison of self-reported and database-linked family history of cancer data in a case-control study. Am J Epidemiol 146 (3): 244-8, 1997.
- Kerr B, Foulkes WD, Cade D, et al.: False family history of breast cancer in the family cancer clinic. Eur J Surg Oncol 24 (4): 275-9, 1998.
- Schwartz MD, Peshkin BN, Hughes C, et al.: Impact of BRCA1/BRCA2 mutation testing on psychologic distress in a clinic-based sample. J Clin Oncol 20 (2): 514-20, 2002.
- Mancini J, Noguès C, Adenis C, et al.: Impact of an information booklet on satisfaction and decision-making about BRCA genetic testing. Eur J Cancer 42 (7): 871-81, 2006.
- Green MJ, Biesecker BB, McInerney AM, et al.: An interactive computer program can effectively educate patients about genetic testing for breast cancer susceptibility. Am J Med Genet 103 (1): 16-23, 2001.
- Green MJ, Peterson SK, Baker MW, et al.: Effect of a computer-based decision aid on knowledge, perceptions, and intentions about genetic testing for breast cancer susceptibility: a randomized controlled trial. JAMA 292 (4): 442-52, 2004.
- Green MJ, McInerney AM, Biesecker BB, et al.: Education about genetic testing for breast cancer susceptibility: patient preferences for a computer program or genetic counselor. Am J Med Genet 103 (1): 24-31, 2001.
- Dabney MK, Huelsman K: Counseling by computer: breast cancer risk and genetic testing. Developed by the University of Wisconsin-Madison Department of Medicine and the Program in Medical Ethics. Genet Test 4 (1): 43-4, 2000.
- Green MJ, Peterson SK, Baker MW, et al.: Use of an educational computer program before genetic counseling for breast cancer susceptibility: effects on duration and content of counseling sessions. Genet Med 7 (4): 221-9, 2005.
- Wang C, Gonzalez R, Milliron KJ, et al.: Genetic counseling for BRCA1/2: a randomized controlled trial of two strategies to facilitate the education and counseling process. Am J Med Genet A 134 (1): 66-73, 2005.
- Joseph G, Beattie MS, Lee R, et al.: Pre-counseling education for low literacy women at risk of Hereditary Breast and Ovarian Cancer (HBOC): patient experiences using the Cancer Risk Education Intervention Tool (CREdIT). J Genet Couns 19 (5): 447-62, 2010.
- Albada A, van Dulmen S, Lindhout D, et al.: A pre-visit tailored website enhances counselees' realistic expectations and knowledge and fulfils information needs for breast cancer genetic counselling. Fam Cancer 11 (1): 85-95, 2012.
- Baty BJ, Kinney AY, Ellis SM: Developing culturally sensitive cancer genetics communication aids for African Americans. Am J Med Genet 118A (2): 146-55, 2003.
- Permuth-Wey J, Vadaparampil S, Rumphs A, et al.: Development of a culturally tailored genetic counseling booklet about hereditary breast and ovarian cancer for Black women. Am J Med Genet A 152A (4): 836-45, 2010.
- Pal T, Stowe C, Cole A, et al.: Evaluation of phone-based genetic counselling in African American women using culturally tailored visual aids. Clin Genet 78 (2): 124-31, 2010.
- Calzone KA: Predisposition testing for breast and ovarian cancer susceptibility. Semin Oncol Nurs 13 (2): 82-90, 1997.
- Smith KR, West JA, Croyle RT, et al.: Familial context of genetic testing for cancer susceptibility: moderating effect of siblings' test results on psychological distress one to two weeks after BRCA1 mutation testing. Cancer Epidemiol Biomarkers Prev 8 (4 Pt 2): 385-92, 1999.
- Wylie JE, Smith KR, Botkin JR: Effects of spouses on distress experienced by BRCA1 mutation carriers over time. Am J Med Genet 119C (1): 35-44, 2003.
- Kash KM, Holland JC, Halper MS, et al.: Psychological distress and surveillance behaviors of women with a family history of breast cancer. J Natl Cancer Inst 84 (1): 24-30, 1992.
- Lerman C, Schwartz M: Adherence and psychological adjustment among women at high risk for breast cancer. Breast Cancer Res Treat 28 (2): 145-55, 1993.
- Kelly PT: Understanding Breast Cancer Risk. Temple University Press, 1991.
- Baty BJ, Venne VL, McDonald J, et al.: BRCA1 testing: genetic counseling protocol development and counseling issues. J Genet Couns 6 (2): 223-44, 1997.
- Richards MP, Hallowell N, Green JM, et al.: Counseling families with hereditary breast and ovarian cancer: a psychosocial perspective. J Genet Couns 4 (3): 219-33, 1995.
- Hoskins KF, Stopfer JE, Calzone KA, et al.: Assessment and counseling for women with a family history of breast cancer. A guide for clinicians. JAMA 273 (7): 577-85, 1995.
- Schneider KA: Genetic counseling for BRCA1/BRCA2 testing. Genet Test 1 (2): 91-8, 1997.
- McKinnon WC, Baty BJ, Bennett RL, et al.: Predisposition genetic testing for late-onset disorders in adults. A position paper of the National Society of Genetic Counselors. JAMA 278 (15): 1217-20, 1997.
- Cummings S, Olopade O: Predisposition testing for inherited breast cancer. Oncology (Huntingt) 12 (8): 1227-41; discussion 1241-2, 1998.
- Lipkus IM, Klein WM, Rimer BK: Communicating breast cancer risks to women using different formats. Cancer Epidemiol Biomarkers Prev 10 (8): 895-8, 2001.
- Butow PN, Lobb EA: Analyzing the process and content of genetic counseling in familial breast cancer consultations. J Genet Couns 13 (5): 403-24, 2004.
- Lerman C, Audrain J, Croyle RT: DNA-testing for heritable breast cancer risks: lessons from traditional genetic counseling. Ann Behav Med 16(4): 327-333, 1994.
- Pieterse AH, van Dulmen AM, Beemer FA, et al.: Cancer genetic counseling: communication and counselees' post-visit satisfaction, cognitions, anxiety, and needs fulfillment. J Genet Couns 16 (1): 85-96, 2007.
- Hamilton JG, Lobel M, Moyer A: Emotional distress following genetic testing for hereditary breast and ovarian cancer: a meta-analytic review. Health Psychol 28 (4): 510-8, 2009.
- Beran TM, Stanton AL, Kwan L, et al.: The trajectory of psychological impact in BRCA1/2 genetic testing: does time heal? Ann Behav Med 36 (2): 107-16, 2008.
- Bosch N, Junyent N, Gadea N, et al.: What factors may influence psychological well being at three months and one year post BRCA genetic result disclosure? Breast 21 (6): 755-60, 2012.
- O'Neill SC, Rini C, Goldsmith RE, et al.: Distress among women receiving uninformative BRCA1/2 results: 12-month outcomes. Psychooncology 18 (10): 1088-96, 2009.
- Oberguggenberger A, Sztankay M, Morscher RJ, et al.: Psychosocial outcomes and counselee satisfaction following genetic counseling for hereditary breast and ovarian cancer: A patient-reported outcome study. J Psychosom Res 89: 39-45, 2016.
- Foster C, Watson M, Eeles R, et al.: Predictive genetic testing for BRCA1/2 in a UK clinical cohort: three-year follow-up. Br J Cancer 96 (5): 718-24, 2007.
- Halbert CH, Stopfer JE, McDonald J, et al.: Long-term reactions to genetic testing for BRCA1 and BRCA2 mutations: does time heal women's concerns? J Clin Oncol 29 (32): 4302-6, 2011.
- Graves KD, Vegella P, Poggi EA, et al.: Long-term psychosocial outcomes of BRCA1/BRCA2 testing: differences across affected status and risk-reducing surgery choice. Cancer Epidemiol Biomarkers Prev 21 (3): 445-55, 2012.
- Cella D, Hughes C, Peterman A, et al.: A brief assessment of concerns associated with genetic testing for cancer: the Multidimensional Impact of Cancer Risk Assessment (MICRA) questionnaire. Health Psychol 21 (6): 564-72, 2002.
- Shkedi-Rafid S, Gabai-Kapara E, Grinshpun-Cohen J, et al.: BRCA genetic testing of individuals from families with low prevalence of cancer: experiences of carriers and implications for population screening. Genet Med 14 (7): 688-94, 2012.
- Metcalfe KA, Poll A, Llacuachaqui M, et al.: Patient satisfaction and cancer-related distress among unselected Jewish women undergoing genetic testing for BRCA1 and BRCA2. Clin Genet 78 (5): 411-7, 2010.
- Metcalfe KA, Mian N, Enmore M, et al.: Long-term follow-up of Jewish women with a BRCA1 and BRCA2 mutation who underwent population genetic screening. Breast Cancer Res Treat 133 (2): 735-40, 2012.
- Armstrong J, Toscano M, Kotchko N, et al.: Utilization and Outcomes of BRCA Genetic Testing and Counseling in a National Commercially Insured Population: The ABOUT Study. JAMA Oncol 1 (9): 1251-60, 2015.
- Dohany L, Gustafson S, Ducaine W, et al.: Psychological distress with direct-to-consumer genetic testing: a case report of an unexpected BRCA positive test result. J Genet Couns 21 (3): 399-401, 2012.
- Mahon SM: Impact of direct-to-consumer genetic testing. J Oncol Pract 8 (4): 260, 2012.
- Jeffers L, Morrison PJ, McCaughan E, et al.: Maximising survival: the main concern of women with hereditary breast and ovarian cancer who undergo genetic testing for BRCA1/2. Eur J Oncol Nurs 18 (4): 411-8, 2014.
- Francke U, Dijamco C, Kiefer AK, et al.: Dealing with the unexpected: consumer responses to direct-access BRCA mutation testing. PeerJ 1: e8, 2013.
- Meiser B, Quinn VF, Gleeson M, et al.: When knowledge of a heritable gene mutation comes out of the blue: treatment-focused genetic testing in women newly diagnosed with breast cancer. Eur J Hum Genet 24 (11): 1517-1523, 2016.
- Ardern-Jones A, Kenen R, Eeles R: Too much, too soon? Patients and health professionals' views concerning the impact of genetic testing at the time of breast cancer diagnosis in women under the age of 40. Eur J Cancer Care (Engl) 14 (3): 272-81, 2005.
- Wevers MR, Hahn DE, Verhoef S, et al.: Breast cancer genetic counseling after diagnosis but before treatment: a pilot study on treatment consequences and psychological impact. Patient Educ Couns 89 (1): 89-95, 2012.
- Zilliacus E, Meiser B, Gleeson M, et al.: Are we being overly cautious? A qualitative inquiry into the experiences and perceptions of treatment-focused germline BRCA genetic testing amongst women recently diagnosed with breast cancer. Support Care Cancer 20 (11): 2949-58, 2012.
- Tercyak KP, Peshkin BN, Brogan BM, et al.: Quality of life after contralateral prophylactic mastectomy in newly diagnosed high-risk breast cancer patients who underwent BRCA1/2 gene testing. J Clin Oncol 25 (3): 285-91, 2007.
- Metcalfe KA, Eisen A, Wright F, et al.: Impact of rapid genetic testing for BRCA1 and BRCA2 at time of breast cancer diagnosis on psychosocial functioning. Breast Cancer Res Treat 191 (3): 631-641, 2022.
- Wevers MR, Ausems MG, Verhoef S, et al.: Does rapid genetic counseling and testing in newly diagnosed breast cancer patients cause additional psychosocial distress? results from a randomized clinical trial. Genet Med 18 (2): 137-44, 2016.
- Wevers MR, Aaronson NK, Bleiker EMA, et al.: Rapid genetic counseling and testing in newly diagnosed breast cancer: Patients' and health professionals' attitudes, experiences, and evaluation of effects on treatment decision making. J Surg Oncol 116 (8): 1029-1039, 2017.
- Conley CC, Ketcher D, Reblin M, et al.: The big reveal: Family disclosure patterns of BRCA genetic test results among young Black women with invasive breast cancer. J Genet Couns 29 (3): 410-422, 2020.
- Patenaude AF, Dorval M, DiGianni LS, et al.: Sharing BRCA1/2 test results with first-degree relatives: factors predicting who women tell. J Clin Oncol 24 (4): 700-6, 2006.
- Lerman C, Peshkin BN, Hughes C, et al.: Family disclosure in genetic testing for cancer susceptibility: determinants and consequences. Journal of Health Care Law and Policy 1 (2): 353-73, 1998.
- Elrick A, Ashida S, Ivanovich J, et al.: Psychosocial and Clinical Factors Associated with Family Communication of Cancer Genetic Test Results among Women Diagnosed with Breast Cancer at a Young Age. J Genet Couns 26 (1): 173-181, 2017.
- Finlay E, Stopfer JE, Burlingame E, et al.: Factors determining dissemination of results and uptake of genetic testing in families with known BRCA1/2 mutations. Genet Test 12 (1): 81-91, 2008.
- Hughes C, Lerman C, Schwartz M, et al.: All in the family: evaluation of the process and content of sisters' communication about BRCA1 and BRCA2 genetic test results. Am J Med Genet 107 (2): 143-50, 2002.
- Baars JE, Ausems MG, van Riel E, et al.: Communication Between Breast Cancer Patients Who Received Inconclusive Genetic Test Results and Their Daughters and Sisters Years After Testing. J Genet Couns 25 (3): 461-71, 2016.
- Wagner Costalas J, Itzen M, Malick J, et al.: Communication of BRCA1 and BRCA2 results to at-risk relatives: a cancer risk assessment program's experience. Am J Med Genet 119C (1): 11-8, 2003.
- MacDonald DJ, Sarna L, van Servellen G, et al.: Selection of family members for communication of cancer risk and barriers to this communication before and after genetic cancer risk assessment. Genet Med 9 (5): 275-82, 2007.
- Kenen R, Arden-Jones A, Eeles R: Healthy women from suspected hereditary breast and ovarian cancer families: the significant others in their lives. Eur J Cancer Care (Engl) 13 (2): 169-79, 2004.
- Claes E, Evers-Kiebooms G, Boogaerts A, et al.: Communication with close and distant relatives in the context of genetic testing for hereditary breast and ovarian cancer in cancer patients. Am J Med Genet 116A (1): 11-9, 2003.
- Foster C, Eeles R, Ardern-Jones A, et al.: Juggling roles and expectations: dilemmas faced by women talking to relatives about cancer and genetic testing. Psychol Health 19 (4): 439-55, 2004.
- Kenen R, Arden-Jones A, Eeles R: We are talking, but are they listening? Communication patterns in families with a history of breast/ovarian cancer (HBOC). Psychooncology 13 (5): 335-45, 2004.
- Segal J, Esplen MJ, Toner B, et al.: An investigation of the disclosure process and support needs of BRCA1 and BRCA2 carriers. Am J Med Genet A 125 (3): 267-72, 2004.
- Bradbury AR, Dignam JJ, Ibe CN, et al.: How often do BRCA mutation carriers tell their young children of the family's risk for cancer? A study of parental disclosure of BRCA mutations to minors and young adults. J Clin Oncol 25 (24): 3705-11, 2007.
- Bradbury AR, Patrick-Miller L, Pawlowski K, et al.: Learning of your parent's BRCA mutation during adolescence or early adulthood: a study of offspring experiences. Psychooncology 18 (2): 200-8, 2009.
- Manne S, Audrain J, Schwartz M, et al.: Associations between relationship support and psychological reactions of participants and partners to BRCA1 and BRCA2 testing in a clinic-based sample. Ann Behav Med 28 (3): 211-25, 2004.
- Daly MB, Montgomery S, Bingler R, et al.: Communicating genetic test results within the family: Is it lost in translation? A survey of relatives in the randomized six-step study. Fam Cancer 15 (4): 697-706, 2016.
- McAllister MF, Evans DG, Ormiston W, et al.: Men in breast cancer families: a preliminary qualitative study of awareness and experience. J Med Genet 35 (9): 739-44, 1998.
- Liede A, Metcalfe K, Hanna D, et al.: Evaluation of the needs of male carriers of mutations in BRCA1 or BRCA2 who have undergone genetic counseling. Am J Hum Genet 67 (6): 1494-504, 2000.
- Metcalfe KA, Liede A, Trinkaus M, et al.: Evaluation of the needs of spouses of female carriers of mutations in BRCA1 and BRCA2. Clin Genet 62 (6): 464-9, 2002.
- Mireskandari S, Sherman KA, Meiser B, et al.: Psychological adjustment among partners of women at high risk of developing breast/ovarian cancer. Genet Med 9 (5): 311-20, 2007.
- Sherman KA, Kasparian NA, Mireskandari S: Psychological adjustment among male partners in response to women's breast/ovarian cancer risk: a theoretical review of the literature. Psychooncology 19 (1): 1-11, 2010.
- Daly MB: The impact of social roles on the experience of men in BRCA1/2 families: implications for counseling. J Genet Couns 18 (1): 42-8, 2009.
- DudokdeWit AC, Tibben A, Frets PG, et al.: Males at-risk for the BRCA1 gene, the psychological impact. Psychooncology 5(3): 251-257, 1996.
- Lodder L, Frets PG, Trijsburg RW, et al.: Men at risk of being a mutation carrier for hereditary breast/ovarian cancer: an exploration of attitudes and psychological functioning during genetic testing. Eur J Hum Genet 9 (7): 492-500, 2001.
- Lerman C, Hughes C, Croyle RT, et al.: Prophylactic surgery decisions and surveillance practices one year following BRCA1/2 testing. Prev Med 31 (1): 75-80, 2000.
- Hughes C, Lynch H, Durham C, et al.: Communication of BRCA1/2 Test Results in Hereditary Breast Cancer Families. Cancer Research in Therapy and Control Vol. 8, 1999, pp. 51-59.
- Tercyak KP, Hughes C, Main D, et al.: Parental communication of BRCA1/2 genetic test results to children. Patient Educ Couns 42 (3): 213-24, 2001.
- Tercyak KP, Peshkin BN, DeMarco TA, et al.: Parent-child factors and their effect on communicating BRCA1/2 test results to children. Patient Educ Couns 47 (2): 145-53, 2002.
- McGivern B, Everett J, Yager GG, et al.: Family communication about positive BRCA1 and BRCA2 genetic test results. Genet Med 6 (6): 503-9, 2004 Nov-Dec.
- Bradbury AR, Patrick-Miller L, Schwartz L, et al.: Psychosocial Adjustment in School-age Girls With a Family History of Breast Cancer. Pediatrics 136 (5): 927-37, 2015.
- Bradbury AR, Patrick-Miller L, Schwartz LA, et al.: Psychosocial Adjustment and Perceived Risk Among Adolescent Girls From Families With BRCA1/2 or Breast Cancer History. J Clin Oncol 34 (28): 3409-16, 2016.
- Tercyak KP, Peshkin BN, Demarco TA, et al.: Information needs of mothers regarding communicating BRCA1/2 cancer genetic test results to their children. Genet Test 11 (3): 249-55, 2007.
- Wertz DC: International perspectives. In: Clarke A, ed.: The Genetic Testing of Children. BIOS Scientific, 1998, pp 271-287.
- Benkendorf JL, Reutenauer JE, Hughes CA, et al.: Patients' attitudes about autonomy and confidentiality in genetic testing for breast-ovarian cancer susceptibility. Am J Med Genet 73 (3): 296-303, 1997.
- Staton AD, Kurian AW, Cobb K, et al.: Cancer risk reduction and reproductive concerns in female BRCA1/2 mutation carriers. Fam Cancer 7 (2): 179-86, 2008.
- Friedman LC, Kramer RM: Reproductive issues for women with BRCA mutations. J Natl Cancer Inst Monogr (34): 83-6, 2005.
- Smith KR, Ellington L, Chan AY, et al.: Fertility intentions following testing for a BRCA1 gene mutation. Cancer Epidemiol Biomarkers Prev 13 (5): 733-40, 2004.
- Quinn G, Vadaparampil S, Wilson C, et al.: Attitudes of high-risk women toward preimplantation genetic diagnosis. Fertil Steril 91 (6): 2361-8, 2009.
- Cunniff C; American Academy of Pediatrics Committee on Genetics: Prenatal screening and diagnosis for pediatricians. Pediatrics 114 (3): 889-94, 2004.
- Rappaport VJ: Prenatal diagnosis and genetic screening--integration into prenatal care. Obstet Gynecol Clin North Am 35 (3): 435-58, ix, 2008.
- Baruch S, Kaufman D, Hudson KL: Genetic testing of embryos: practices and perspectives of US in vitro fertilization clinics. Fertil Steril 89 (5): 1053-8, 2008.
- Ogilvie CM, Braude PR, Scriven PN: Preimplantation genetic diagnosis--an overview. J Histochem Cytochem 53 (3): 255-60, 2005.
- Vadaparampil ST, Quinn GP, Knapp C, et al.: Factors associated with preimplantation genetic diagnosis acceptance among women concerned about hereditary breast and ovarian cancer. Genet Med 11 (10): 757-65, 2009.
- Quinn GP, Vadaparampil ST, King LM, et al.: Conflict between values and technology: perceptions of preimplantation genetic diagnosis among women at increased risk for hereditary breast and ovarian cancer. Fam Cancer 8 (4): 441-9, 2009.
- Chan JL, Johnson LNC, Sammel MD, et al.: Reproductive Decision-Making in Women with BRCA1/2 Mutations. J Genet Couns 26 (3): 594-603, 2017.
- Menon U, Harper J, Sharma A, et al.: Views of BRCA gene mutation carriers on preimplantation genetic diagnosis as a reproductive option for hereditary breast and ovarian cancer. Hum Reprod 22 (6): 1573-7, 2007.
- Fortuny D, Balmaña J, Graña B, et al.: Opinion about reproductive decision making among individuals undergoing BRCA1/2 genetic testing in a multicentre Spanish cohort. Hum Reprod 24 (4): 1000-6, 2009.
- Julian-Reynier C, Fabre R, Coupier I, et al.: BRCA1/2 carriers: their childbearing plans and theoretical intentions about having preimplantation genetic diagnosis and prenatal diagnosis. Genet Med 14 (5): 527-34, 2012.
- Ormondroyd E, Donnelly L, Moynihan C, et al.: Attitudes to reproductive genetic testing in women who had a positive BRCA test before having children: a qualitative analysis. Eur J Hum Genet 20 (1): 4-10, 2012.
- Quinn GP, Vadaparampil ST, Miree CA, et al.: High risk men's perceptions of pre-implantation genetic diagnosis for hereditary breast and ovarian cancer. Hum Reprod 25 (10): 2543-50, 2010.
- Struewing JP, Abeliovich D, Peretz T, et al.: The carrier frequency of the BRCA1 185delAG mutation is approximately 1 percent in Ashkenazi Jewish individuals. Nat Genet 11 (2): 198-200, 1995.
- Rothenberg KH: Breast cancer, the genetic "quick fix," and the Jewish community. Ethical, legal, and social challenges. Health Matrix Clevel 7 (1): 97-124, 1997 Winter.
- Foster MW, Bernsten D, Carter TH: A model agreement for genetic research in socially identifiable populations. Am J Hum Genet 63 (3): 696-702, 1998.
- Burhansstipanov L, Bemis LT, Dignan MB: Native American cancer education: genetic and cultural issues. J Cancer Educ 16 (3): 142-5, 2001 Autumn.
- Hughes C, Fasaye GA, LaSalle VH, et al.: Sociocultural influences on participation in genetic risk assessment and testing among African American women. Patient Educ Couns 51 (2): 107-14, 2003.
- Julian-Reynier CM, Bouchard LJ, Evans DG, et al.: Women's attitudes toward preventive strategies for hereditary breast or ovarian carcinoma differ from one country to another: differences among English, French, and Canadian women. Cancer 92 (4): 959-68, 2001.
- Phillips KA, Warner E, Meschino WS, et al.: Perceptions of Ashkenazi Jewish breast cancer patients on genetic testing for mutations in BRCA1 and BRCA2. Clin Genet 57 (5): 376-83, 2000.
- Vadaparampil ST, Quinn GP, Small BJ, et al.: A pilot study of hereditary breast and ovarian knowledge among a multiethnic group of Hispanic women with a personal or family history of cancer. Genet Test Mol Biomarkers 14 (1): 99-106, 2010.
- Halbert CH, Kessler L, Troxel AB, et al.: Effect of genetic counseling and testing for BRCA1 and BRCA2 mutations in African American women: a randomized trial. Public Health Genomics 13 (7-8): 440-8, 2010.
- Freedman TG: Genetic susceptibility testing: ethical and social quandaries. Health Soc Work 23 (3): 214-22, 1998.
- Hubbard R, Lewontin RC: Pitfalls of genetic testing. N Engl J Med 334 (18): 1192-4, 1996.
- Parens E: Glad and terrified: on the ethics of BRACA1 and 2 testing. Cancer Invest 14 (4): 405-11, 1996.
- Winter PR, Wiesner GL, Finnegan J, et al.: Notification of a family history of breast cancer: issues of privacy and confidentiality. Am J Med Genet 66 (1): 1-6, 1996.
- Geller G, Doksum T, Bernhardt BA, et al.: Participation in breast cancer susceptibility testing protocols: influence of recruitment source, altruism, and family involvement on women's decisions. Cancer Epidemiol Biomarkers Prev 8 (4 Pt 2): 377-83, 1999.
- Rimer BK, Schildkraut JM, Lerman C, et al.: Participation in a women's breast cancer risk counseling trial. Who participates? Who declines? High Risk Breast Cancer Consortium. Cancer 77 (11): 2348-55, 1996.
- Robson ME, Storm CD, Weitzel J, et al.: American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol 28 (5): 893-901, 2010.
- Burke W, Daly M, Garber J, et al.: Recommendations for follow-up care of individuals with an inherited predisposition to cancer. II. BRCA1 and BRCA2. Cancer Genetics Studies Consortium. JAMA 277 (12): 997-1003, 1997.
- Durfy SJ, Buchanan TE, Burke W: Testing for inherited susceptibility to breast cancer: a survey of informed consent forms for BRCA1 and BRCA2 mutation testing. Am J Med Genet 75 (1): 82-7, 1998.
- Statement of the American Society of Clinical Oncology: genetic testing for cancer susceptibility, Adopted on February 20, 1996. J Clin Oncol 14 (5): 1730-6; discussion 1737-40, 1996.
- Hallowell N, Foster C, Eeles R, et al.: Balancing autonomy and responsibility: the ethics of generating and disclosing genetic information. J Med Ethics 29 (2): 74-9; discussion 80-3, 2003.
- Krassuski L, Vennedey V, Stock S, et al.: Effectiveness of decision aids for female BRCA1 and BRCA2 mutation carriers: a systematic review. BMC Med Inform Decis Mak 19 (1): 154, 2019.
- Metcalfe KA, Pal T, Narod SA, et al.: Theory-based behavior change intervention to increase uptake of risk-reducing salpingo-oophorectomy in women with a BRCA1 or BRCA2 pathogenic variant: The PREVENT randomized controlled trial. Cancer Med 12 (17): 18246-18257, 2023.
- Metcalfe K, Eisen A, Senter L, et al.: International trends in the uptake of cancer risk reduction strategies in women with a BRCA1 or BRCA2 mutation. Br J Cancer 121 (1): 15-21, 2019.
- Marcinkute R, Woodward ER, Gandhi A, et al.: Uptake and efficacy of bilateral risk reducing surgery in unaffected female BRCA1 and BRCA2 carriers. J Med Genet 59 (2): 133-140, 2022.
- Schwartz MD, Isaacs C, Graves KD, et al.: Long-term outcomes of BRCA1/BRCA2 testing: risk reduction and surveillance. Cancer 118 (2): 510-7, 2012.
- Garcia C, Wendt J, Lyon L, et al.: Risk management options elected by women after testing positive for a BRCA mutation. Gynecol Oncol 132 (2): 428-33, 2014.
- Schwartz MD, Lerman C, Brogan B, et al.: Impact of BRCA1/BRCA2 counseling and testing on newly diagnosed breast cancer patients. J Clin Oncol 22 (10): 1823-9, 2004.
- Armstrong J, Lynch K, Virgo KS, et al.: Utilization, Timing, and Outcomes of BRCA Genetic Testing Among Women With Newly Diagnosed Breast Cancer From a National Commercially Insured Population: The ABOARD Study. JCO Oncol Pract 17 (2): e226-e235, 2021.
- Bradbury AR, Ibe CN, Dignam JJ, et al.: Uptake and timing of bilateral prophylactic salpingo-oophorectomy among BRCA1 and BRCA2 mutation carriers. Genet Med 10 (3): 161-166, 2008.
- Park SY, Kim Y, Kim S: Factors associated with the decision to undergo risk-reducing salpingo-oophorectomy among women at high risk for hereditary breast and ovarian cancer: a systematic review. Korean J Women Health Nurs 26 (4): 285-299, 2020.
- Cortesi L, Razzaboni E, Toss A, et al.: A rapid genetic counselling and testing in newly diagnosed breast cancer is associated with high rate of risk-reducing mastectomy in BRCA1/2-positive Italian women. Ann Oncol 25 (1): 57-63, 2014.
- Rosenberg SM, Ruddy KJ, Tamimi RM, et al.: BRCA1 and BRCA2 Mutation Testing in Young Women With Breast Cancer. JAMA Oncol 2 (6): 730-6, 2016.
- Howard-McNatt M, Schroll RW, Hurt GJ, et al.: Contralateral prophylactic mastectomy in breast cancer patients who test negative for BRCA mutations. Am J Surg 202 (3): 298-302, 2011.
- Perez L, Webster E, Bull L, et al.: Patient perspectives on risk-reducing salpingectomy with delayed oophorectomy for ovarian cancer risk-reduction: A systematic review of the literature. Gynecol Oncol 173: 106-113, 2023.
- Nebgen DR, Hurteau J, Holman LL, et al.: Bilateral salpingectomy with delayed oophorectomy for ovarian cancer risk reduction: A pilot study in women with BRCA1/2 mutations. Gynecol Oncol 150 (1): 79-84, 2018.
- MacDonald DJ, Sarna L, Uman GC, et al.: Cancer screening and risk-reducing behaviors of women seeking genetic cancer risk assessment for breast and ovarian cancers. Oncol Nurs Forum 33 (2): E27-35, 2006.
- Litton JK, Westin SN, Ready K, et al.: Perception of screening and risk reduction surgeries in patients tested for a BRCA deleterious mutation. Cancer 115 (8): 1598-604, 2009.
- Tyndel S, Austoker J, Henderson BJ, et al.: What is the psychological impact of mammographic screening on younger women with a family history of breast cancer? Findings from a prospective cohort study by the PIMMS Management Group. J Clin Oncol 25 (25): 3823-30, 2007.
- Rees G, Young MA, Gaff C, et al.: A qualitative study of health professionals' views regarding provision of information about health-protective behaviors during genetic consultation for breast cancer. J Genet Couns 15 (2): 95-104, 2006.
- Lodder LN, Frets PG, Trijsburg RW, et al.: One year follow-up of women opting for presymptomatic testing for BRCA1 and BRCA2: emotional impact of the test outcome and decisions on risk management (surveillance or prophylactic surgery). Breast Cancer Res Treat 73 (2): 97-112, 2002.
- van Oostrom I, Meijers-Heijboer H, Lodder LN, et al.: Long-term psychological impact of carrying a BRCA1/2 mutation and prophylactic surgery: a 5-year follow-up study. J Clin Oncol 21 (20): 3867-74, 2003.
- Bresser PJ, Seynaeve C, Van Gool AR, et al.: The course of distress in women at increased risk of breast and ovarian cancer due to an (identified) genetic susceptibility who opt for prophylactic mastectomy and/or salpingo-oophorectomy. Eur J Cancer 43 (1): 95-103, 2007.
- Frost MH, Schaid DJ, Sellers TA, et al.: Long-term satisfaction and psychological and social function following bilateral prophylactic mastectomy. JAMA 284 (3): 319-24, 2000.
- Metcalfe KA, Esplen MJ, Goel V, et al.: Psychosocial functioning in women who have undergone bilateral prophylactic mastectomy. Psychooncology 13 (1): 14-25, 2004.
- Schlich-Bakker KJ, Ausems MG, Schipper M, et al.: BRCA1/2 mutation testing in breast cancer patients: a prospective study of the long-term psychological impact of approach during adjuvant radiotherapy. Breast Cancer Res Treat 109 (3): 507-14, 2008.
- Frost MH, Slezak JM, Tran NV, et al.: Satisfaction after contralateral prophylactic mastectomy: the significance of mastectomy type, reconstructive complications, and body appearance. J Clin Oncol 23 (31): 7849-56, 2005.
- Schwartz MD: Contralateral prophylactic mastectomy: efficacy, satisfaction, and regret. J Clin Oncol 23 (31): 7777-9, 2005.
- Metcalfe KA, Cil TD, Semple JL, et al.: Long-Term Psychosocial Functioning in Women with Bilateral Prophylactic Mastectomy: Does Preservation of the Nipple-Areolar Complex Make a Difference? Ann Surg Oncol 22 (10): 3324-30, 2015.
- Yao K, Liederbach E, Tang R, et al.: Nipple-sparing mastectomy in BRCA1/2 mutation carriers: an interim analysis and review of the literature. Ann Surg Oncol 22 (2): 370-6, 2015.
- Geiger AM, Nekhlyudov L, Herrinton LJ, et al.: Quality of life after bilateral prophylactic mastectomy. Ann Surg Oncol 14 (2): 686-94, 2007.
- Isern AE, Tengrup I, Loman N, et al.: Aesthetic outcome, patient satisfaction, and health-related quality of life in women at high risk undergoing prophylactic mastectomy and immediate breast reconstruction. J Plast Reconstr Aesthet Surg 61 (10): 1177-87, 2008.
- Kenen RH, Shapiro PJ, Hantsoo L, et al.: Women with BRCA1 or BRCA2 mutations renegotiating a post-prophylactic mastectomy identity: self-image and self-disclosure. J Genet Couns 16 (6): 789-98, 2007.
- Altschuler A, Nekhlyudov L, Rolnick SJ, et al.: Positive, negative, and disparate--women's differing long-term psychosocial experiences of bilateral or contralateral prophylactic mastectomy. Breast J 14 (1): 25-32, 2008 Jan-Feb.
- Patenaude AF, Orozco S, Li X, et al.: Support needs and acceptability of psychological and peer consultation: attitudes of 108 women who had undergone or were considering prophylactic mastectomy. Psychooncology 17 (8): 831-43, 2008.
- Hall E, Finch A, Jacobson M, et al.: Effects of bilateral salpingo-oophorectomy on menopausal symptoms and sexual functioning among women with a BRCA1 or BRCA2 mutation. Gynecol Oncol 152 (1): 145-150, 2019.
- Powell CB, Alabaster A, Le A, et al.: Sexual function, menopausal symptoms, depression and cancer worry in women with BRCA mutations. Psychooncology 29 (2): 331-338, 2020.
- Terra L, Lee Meeuw Kjoe PR, Agelink van Rentergem JA, et al.: Long-term effects of premenopausal risk-reducing salpingo-oophorectomy on cognition in women with high familial risk of ovarian cancer: A cross-sectional study. BJOG 130 (8): 968-977, 2023.
- Isaacs C, Peshkin BN, Schwartz M, et al.: Breast and ovarian cancer screening practices in healthy women with a strong family history of breast or ovarian cancer. Breast Cancer Res Treat 71 (2): 103-12, 2002.
- Peshkin BN, Schwartz MD, Isaacs C, et al.: Utilization of breast cancer screening in a clinically based sample of women after BRCA1/2 testing. Cancer Epidemiol Biomarkers Prev 11 (10 Pt 1): 1115-8, 2002.
- Tinley ST, Houfek J, Watson P, et al.: Screening adherence in BRCA1/2 families is associated with primary physicians' behavior. Am J Med Genet A 125 (1): 5-11, 2004.
- Lerman C, Seay J, Balshem A, et al.: Interest in genetic testing among first-degree relatives of breast cancer patients. Am J Med Genet 57 (3): 385-92, 1995.
- Struewing JP, Lerman C, Kase RG, et al.: Anticipated uptake and impact of genetic testing in hereditary breast and ovarian cancer families. Cancer Epidemiol Biomarkers Prev 4 (2): 169-73, 1995.
- Jacobsen PB, Valdimarsdottier HB, Brown KL, et al.: Decision-making about genetic testing among women at familial risk for breast cancer. Psychosom Med 59 (5): 459-66, 1997 Sep-Oct.
- Watson M, Kash KM, Homewood J, et al.: Does genetic counseling have any impact on management of breast cancer risk? Genet Test 9 (2): 167-74, 2005.
Latest Updates to This Summary (04/04/2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Introduction
Updated statistics with estimated new cancer cases and deaths for 2024 (cited American Cancer Society as reference 1).
Psychosocial Issues in Hereditary Breast and Gynecologic Cancers
Added text to state that a systematic review of decision aids for BRCA1/BRCA2 carriers suggested that these tools are beneficial during the decision-making process (cited Krassuski et al. as reference 216). Also added text to state that BRCA1/BRCA2 carriers who used decision aids had lower decisional conflict, were more likely to reach risk-management decisions, and were more satisfied with their decisions than those who did not use decision aids. There were mixed or null findings regarding information-related outcomes like participant knowledge and whether there were consistent differences in actual risk-management choices.
Added text to state that a telephone-based intervention study used integrated motivational interviewing techniques and was led by genetic counselors (cited Metcalfe et al. as reference 217). Also added text to state that this study found no differences in rates of risk-reducing salpingo-oophorectomy (RRSO) among BRCA1/BRCA2 carriers in the intervention arm and the control arm after 1 year. Women in the intervention arm had significantly lower decisional conflict and higher knowledge scores after 1 year. Two years after the intervention, 53.9% of women in the intervention arm had undergone RRSO, while 32.6% of women in the control arm had undergone RRSO.
Added text to state that RRSO was associated with a personal history of mastectomy, higher perceived risk of ovarian cancer, and higher perceived benefit of RRSO (cited Park et al. as reference 225).
Added text to state that a systematic review of risk-reducing salpingectomy with delayed oophorectomy (RRDSO) found that the avoidance of surgical menopause, preservation of fertility, concerns about sexual functioning, a family history of breast cancer, and an interest to avoid hormone replacement therapy were all related to being more likely to accept this option (cited Perez et al. as reference 229). Also added text to state that those concerned with surgical timing, complications, and ongoing cancer risk were less likely to opt for RRDSO. In a small cohort of premenopausal BRCA1/BRCA2 carriers, 44% chose to pursue RRDSO, 28% chose to pursue RRSO, and 28% chose to pursue only screening (cited Nebgen et al. as reference 230). Also added text to state that 1 year after surgery, cancer worry had decreased among all of those who elected to have surgery. Surgical complications were not reported.
The Psychosocial outcomes associated with RRSO subsection was extensively revised.
This summary is written and maintained by the PDQ Cancer Genetics Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the genetics of breast and gynecologic cancers. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Cancer Genetics Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Genetics of Breast and Gynecologic Cancers are:
- Doreen Agnese, MD (The Ohio State University)
- Kathleen A. Calzone, PhD, RN, AGN-BC, FAAN (National Cancer Institute)
- Ilana Cass, MD (Dartmouth-Hitchcock Medical Center)
- Lee-may Chen, MD (UCSF Helen Diller Family Comprehensive Cancer Center)
- Mary B. Daly, MD, PhD (Fox Chase Cancer Center)
- Megan Frone, MS, CGC (National Cancer Institute)
- Joanne Kotsopoulos, PhD (University of Toronto and Women's College Hospital)
- Suzanne C. O'Neill, PhD (Georgetown University)
- Tuya Pal, MD, FACMG, FCCMG (Vanderbilt-Ingram Cancer Center)
- Susan K. Peterson, PhD, MPH (University of Texas, M.D. Anderson Cancer Center)
- John M. Quillin, PhD, MPH, MS (Virginia Commonwealth University)
- Padma Sheila Rajagopal, MD, MPH, MSC (National Cancer Institute)
- Charite Ricker, MS, CGC (University of Southern California)
- Mary Beth Terry, PhD (Columbia University Mailman School of Public Health)
- Catharine Wang, PhD, MSc (Boston University School of Public Health)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Cancer Genetics Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Cancer Genetics Editorial Board. PDQ Genetics of Breast and Gynecologic Cancers. Bethesda, MD: National Cancer Institute. Updated
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
The information in these summaries should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.