National Cancer Institute
Post Date: Apr 4, 2024
Evidence-based, expert-reviewed summary about the von Hippel-Lindau disease (VHL), including information about the VHL gene. This summary also contains information about screening for VHL, clinical presentation, management, and prognosis of VHL.
Von Hippel-Lindau Disease
Introduction
Von Hippel-Lindau disease (VHL) is an autosomal dominant disease that can predispose individuals to multiple neoplasms. Germlinepathogenic variants in the VHLgene predispose individuals to specific types of benign tumors, malignant tumors, and cysts in many organ systems. These tumors and cysts include central nervous system hemangioblastomas; retinal hemangioblastomas; clear cell renal cell carcinomas and renal cysts; pheochromocytomas, cysts, cystadenomas, and neuroendocrine tumors of the pancreas; endolymphatic sac tumors; and cystadenomas of the epididymis (males) and of the broad ligament (females). A multidisciplinary approach is required for the evaluation, and in some cases, the management of individuals with VHL. A case manager or nurse navigator may be helpful in certain scenarios. Specialists who care for individuals with VHL may include urologic oncology surgeons, neurosurgeons, general surgeons, ophthalmologists, endocrinologists, neurologists, medical oncologists, genetic counselors, and medical geneticists.
References
- Choyke PL, Glenn GM, Walther MM, et al.: von Hippel-Lindau disease: genetic, clinical, and imaging features. Radiology 194 (3): 629-42, 1995.
- Lonser RR, Glenn GM, Walther M, et al.: von Hippel-Lindau disease. Lancet 361 (9374): 2059-67, 2003.
- Pithukpakorn M, Glenn G: von Hippel-Lindau syndrome. Community Oncology 1 (4): 232-43, 2004.
- Glenn GM, Daniel LN, Choyke P, et al.: Von Hippel-Lindau (VHL) disease: distinct phenotypes suggest more than one mutant allele at the VHL locus. Hum Genet 87 (2): 207-10, 1991.
- Wolters WPG, Dreijerink KMA, Giles RH, et al.: Multidisciplinary integrated care pathway for von Hippel-Lindau disease. Cancer 128 (15): 2871-2879, 2022.
Genetics
VHL Gene
The VHLgene is a tumor suppressor gene located on the short arm of chromosome 3 at cytoband 3p25-26. VHLpathogenic variants occur in all three exons of this gene. Most affected individuals inherit a VHLgermline pathogenic variant from an affected parent and a normal (wild-type) copy of VHL from an unaffected parent. Von Hippel-Lindau disease (VHL)-associated tumors conform to Knudson’s “two-hit” hypothesis, in which the clonal origin, or the first transformed cell of the tumor, occurs only after both VHLalleles are inactivated in a cell. The inherited germline pathogenic variant in VHL represents the first "hit," which is present in every cell in the body. The second “hit” is a somatic variant, one that occurs in a specific tissue at some point after a person's birth. It damages the normal, or wild-type, VHL allele, creating a clonal neoplastic cell of origin, which may proliferate into a tumor mass.
Prevalence and rare founder effects
The incidence of VHL is estimated to be between 1 case per 27,000 and 1 case per 43,000 live births in the general population. The prevalence is estimated to be between 1 in 31,000 to 1 in 91,000 individuals. Precise quantification of this number is a challenge because it requires comprehensive screening of potentially at-risk blood relatives of individuals diagnosed with VHL. The large number of unique pathogenic variants in this small three-exon gene indicates that most family clusters have not arisen from a single founder.
Penetrance of pathogenic variants
VHL pathogenic variants are highly penetrant, with manifestations found in more than 90% of carriers by age 65 years. Almost all carriers develop one or more types of VHL-related neoplasms.
Risk factors for VHL
Each offspring of an individual with VHL has a 50% chance of inheriting the VHL pathogenic variant allele from their affected parent. For more information, see the Genetic Diagnosis in VHL section.
Genotype-phenotype correlations
Specific alterations in the VHL gene may help predict an individual’s risk of developing renal cell carcinoma (RCC) and other VHL-associated tumors. Classifying genotypes into different risk groups has been a goal to better inform screening for VHL disease manifestations. There have been efforts to subdivide patients based on familial variants. For example, in 1991, researchers classified VHL cases into two different types: type 1 VHL (VHL without pheochromocytomas [PHEOs]) and type 2 VHL (VHL with PHEOs). Individuals with type 1 VHL often have large deletions in the VHL gene, whereas individuals with type 2 VHL often have missense variants in the VHL gene. In 1995, type 2 VHL was further subdivided into type 2A VHL (VHL with PHEOs and a low risk for RCC) and type 2B VHL (VHL with PHEOs and RCC). In 2001, type 2C VHL (VHL with isolated PHEOs but without hemangioblastoma or RCC) was also reported. The following types of pathogenic variants can lead to VHL clinical manifestations: missense variants, nonsense variants, frameshift variants, insertions, partial/complete deletions, and splice-site variants. These can be further divided into truncating and nontruncating variants.
In recent years, there have been exceptions to these VHL classifications, suggesting that screening for all disease manifestations is warranted in all individuals with VHL. Given the considerable overlap between VHL phenotypes, current surveillance guidelines do not incorporate an individual's genotype or clinical VHL subtype into screening recommendations.
De novo pathogenic variants and mosaicism
In some cases, an individual can be diagnosed with VHL, even when this disease is not present in other family members. This scenario can occur when an affected individual has a de novo (new) pathogenic variant in the VHL gene. Patients who were diagnosed with VHL and have no family history of VHL comprise about 23% of VHL kindreds. A new variant is, by definition, a postzygotic event because it is not transmitted from a parent.
Depending on the embryogenesis stage at which the new variant occurs, there may be different somatic cell lineages carrying the variant. This influences the extent of mosaicism seen in the cell lineages. Mosaicism occurs when two or more cell lines in an individual differ by genotype. These differing cell lines all arise from the same zygote. If the postzygotic de novo variant affects the gonadal cell line, there is a risk of transmitting a germline variant to offspring.
Allelic disorder
VHL-associated polycythemia (also known as familial erythrocytosis type 2 or Chuvash polycythemia) is a rare, autosomal recessive blood disorder caused by homozygous or compound heterozygous pathogenic variants in VHL in which affected individuals develop abnormally high numbers of red blood cells (polycythemia). The affected individuals have biallelic pathogenic variants in the VHL gene. It had been originally thought that the typical VHL syndromic tumors do not occur in these affected individuals.
Other Genetic Alterations
In sporadic RCC, mutational inactivation of the VHL gene is the most frequent molecular event. In addition to VHL inactivation, sporadic clear cell renal cell carcinoma (ccRCC) tumors harbor frequent variants in other genes, including PBRM1, SETD2, and BAP1. Mutational inactivation of PBRM1, SETD2, and BAP1 are “second hit” events that occur after VHL is altered in sporadic ccRCC. These events contribute to the development and growth of ccRCC. Germline pathogenic variants in PBRM1 and BAP1 can result in hereditary forms of ccRCC. The role of PBRM1, BAP1, and SETD2 in VHL-related ccRCC growth and progression is under investigation.
References
- Latif F, Tory K, Gnarra J, et al.: Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 260 (5112): 1317-20, 1993.
- Knudson AG, Strong LC: Mutation and cancer: neuroblastoma and pheochromocytoma. Am J Hum Genet 24 (5): 514-32, 1972.
- Knudson AG: Genetics of human cancer. Annu Rev Genet 20: 231-51, 1986.
- Maher ER, Iselius L, Yates JR, et al.: Von Hippel-Lindau disease: a genetic study. J Med Genet 28 (7): 443-7, 1991.
- Binderup ML, Galanakis M, Budtz-Jørgensen E, et al.: Prevalence, birth incidence, and penetrance of von Hippel-Lindau disease (vHL) in Denmark. Eur J Hum Genet 25 (3): 301-307, 2017.
- Evans DG, Howard E, Giblin C, et al.: Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A 152A (2): 327-32, 2010.
- Neumann HP, Wiestler OD: Clustering of features of von Hippel-Lindau syndrome: evidence for a complex genetic locus. Lancet 337 (8749): 1052-4, 1991.
- Poulsen ML, Budtz-Jørgensen E, Bisgaard ML: Surveillance in von Hippel-Lindau disease (vHL). Clin Genet 77 (1): 49-59, 2010.
- Reich M, Jaegle S, Neumann-Haefelin E, et al.: Genotype-phenotype correlation in von Hippel-Lindau disease. Acta Ophthalmol 99 (8): e1492-e1500, 2021.
- Salama Y, Albanyan S, Szybowska M, et al.: Comprehensive characterization of a Canadian cohort of von Hippel-Lindau disease patients. Clin Genet 96 (5): 461-467, 2019.
- Brauch H, Kishida T, Glavac D, et al.: Von Hippel-Lindau (VHL) disease with pheochromocytoma in the Black Forest region of Germany: evidence for a founder effect. Hum Genet 95 (5): 551-6, 1995.
- Hoffman MA, Ohh M, Yang H, et al.: von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet 10 (10): 1019-27, 2001.
- Sgambati MT, Stolle C, Choyke PL, et al.: Mosaicism in von Hippel-Lindau disease: lessons from kindreds with germline mutations identified in offspring with mosaic parents. Am J Hum Genet 66 (1): 84-91, 2000.
- Austin KD, Hall JG: Nontraditional inheritance. Pediatr Clin North Am 39 (2): 335-48, 1992.
- Ang SO, Chen H, Hirota K, et al.: Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet 32 (4): 614-21, 2002.
- Pastore YD, Jelinek J, Ang S, et al.: Mutations in the VHL gene in sporadic apparently congenital polycythemia. Blood 101 (4): 1591-5, 2003.
- Cario H, Schwarz K, Jorch N, et al.: Mutations in the von Hippel-Lindau (VHL) tumor suppressor gene and VHL-haplotype analysis in patients with presumable congenital erythrocytosis. Haematologica 90 (1): 19-24, 2005.
- Popova T, Hebert L, Jacquemin V, et al.: Germline BAP1 mutations predispose to renal cell carcinomas. Am J Hum Genet 92 (6): 974-80, 2013.
- Farley MN, Schmidt LS, Mester JL, et al.: A novel germline mutation in BAP1 predisposes to familial clear-cell renal cell carcinoma. Mol Cancer Res 11 (9): 1061-71, 2013.
- Cancer Genome Atlas Research Network: Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499 (7456): 43-9, 2013.
- Benusiglio PR, Couvé S, Gilbert-Dussardier B, et al.: A germline mutation in PBRM1 predisposes to renal cell carcinoma. J Med Genet 52 (6): 426-30, 2015.
Molecular Biology
The VHLtumor suppressor gene encodes two proteins: a 213 amino acid protein (pVHL30) and a 154 amino acid protein, which is the product of internal translation. pVHL is best known for its regulation of hypoxia-inducible factor (HIF) activity, which is linked to tumor suppression. Other reported functions of pVHL include regulation of extracellular matrix formation, microtubule and centrosome maturation, and p53 inactivation. These functions are described in more detail in the following paragraphs.
HIF1-Alpha and HIF2-Alpha
pVHL regulates protein levels of HIF1-alpha and HIF2-alpha in the cell by acting as a substrate recognition site for HIF as part of an E3 ubiquitin ligase complex. In normoxic conditions, HIF1-alpha and HIF2-alpha are enzymatically hydroxylated by intracellular prolyl hydroxylases. The hydroxylated HIF subunits are bound by the VHL protein complex, covalently linked to ubiquitin, and degraded by the S26 proteasome.
Hypoxia inactivates prolyl hydroxylases, leading to lack of HIF hydroxylation. Nonhydroxylated HIF1-alpha and HIF2-alpha are not bound to the VHL protein complex for ubiquitination, and therefore, accumulate. The resulting constitutively high levels of HIF1-alpha and HIF2-alpha drive increased transcription of a variety of genes, including growth and angiogenic factors, enzymes of the intermediary metabolism, and genes promoting stemness-like cellular phenotypes.
HIF1-alpha and HIF2-alpha possess distinct and partially contrasting functional characteristics. In the context of renal cell carcinoma (RCC), it appears that the EPAS1 gene, also known as HIF2A, acts as an oncogene, and HIF1A acts as a tumor suppressor gene. HIF2-alpha may preferentially upregulate Myc activity, whereas HIF1-alpha may inhibit Myc activity. Hypoxia-associated factor has been shown to increase HIF2-alpha transactivation and HIF1-alpha instability. Preferential loss of chromosome 14q, the locus for the HIF1A gene, results in decreased levels of HIF1-alpha protein.
Numerous studies using xenografted or transgenic animal models have shown that inactivation of HIF2-alpha by pVHL is necessary and sufficient for tumor suppression by the pVHL proteins. HIF2-alpha is now an established therapeutic target for von Hippel-Lindau disease (VHL)-related malignancies. Specific HIF2-alpha inhibitors are in preclinical and clinical testing.
Microtubule Regulation and Cilia Centrosome Control
Emerging data point to the importance of pVHL-mediated control of the primary cilium and the cilia centrosome cycle. The nonmotile primary cilium acts as a mechanosensor, regulates cell signaling, and controls cellular entry into mitosis. The loss of primary ciliary function results in the loss of the cell’s ability to maintain planar cell polarity; thus ultimately results in cyst formation. The loss of pVHL results in the loss of the primary cilium. pVHL binds to and stabilizes microtubules in a glycogen synthase 3–dependent fashion. The loss of pVHL, or expression of variant pVHL in cells, also results in unstable astral microtubules, dysregulation of the spindle assembly checkpoint, and an increase in aneuploidy.
Cell Cycle Control
pVHL reintroduction induces cell cycle arrest and p27 upregulation after serum withdrawal in VHL-null cell lines. Additionally, pVHL destabilizes Skp2, and upregulates p27 in response to DNA damage. Nuclear localization and intensity of p27 is inversely associated with tumor grade. pVHL binds to and facilitates phosphorylation of p53 in an ATM-dependent fashion.
Extracellular Matrix Control
Functional pVHL is needed for appropriate assembly of an extracellular fibronectin matrix. Additionally, phosphorylation of pVHL regulates binding of fibronectin and secretion into the extracellular space.
Regulation of Oncogenic Autophagy
In clear cell renal cell carcinoma (ccRCC), oncogenic autophagy dependent on microtubule-associated protein 1 light chain 3 alpha and beta (LC3A and LC3B) is stimulated by activity of the transient receptor potential melastatin 3 (TRPM3) channel through multiple complementary mechanisms. The VHL tumor suppressor represses this oncogenic autophagy in a coordinated manner through the activity of miR-204, which is expressed from intron 6 of the gene encoding TRPM3. TRPM3 represents an actionable target for ccRCC treatment.
Animal Models of VHL
Vhl-knockout mice die in utero. HeterozygousVhl mice develop vascular liver lesions reminiscent of hemangioblastomas. Conditional targeted inactivation of the Vhl gene in the mouse kidney results in the generation of VHL-resembling cysts but not RCC. Coordinate inactivation of Vhl and Pten results in a higher rate of cyst formation, but no obvious RCC. Murine homologues of the Vhl R200W pathogenic variant induced polycythemia in mice, phenocopying Chuvash polycythemia. The discovery of several new potential tumor suppressor genes inactivated in the context of RCC, including PBRM1, SETD2, and BAP1, provide new avenues for developing relevant animal models of at least some VHL manifestations.
References
- Iliopoulos O, Ohh M, Kaelin WG: pVHL19 is a biologically active product of the von Hippel-Lindau gene arising from internal translation initiation. Proc Natl Acad Sci U S A 95 (20): 11661-6, 1998.
- Pause A, Lee S, Lonergan KM, et al.: The von Hippel-Lindau tumor suppressor gene is required for cell cycle exit upon serum withdrawal. Proc Natl Acad Sci U S A 95 (3): 993-8, 1998.
- Kurban G, Hudon V, Duplan E, et al.: Characterization of a von Hippel Lindau pathway involved in extracellular matrix remodeling, cell invasion, and angiogenesis. Cancer Res 66 (3): 1313-9, 2006.
- Thoma CR, Toso A, Gutbrodt KL, et al.: VHL loss causes spindle misorientation and chromosome instability. Nat Cell Biol 11 (8): 994-1001, 2009.
- Maxwell PH, Wiesener MS, Chang GW, et al.: The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399 (6733): 271-5, 1999.
- Ivan M, Kondo K, Yang H, et al.: HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292 (5516): 464-8, 2001.
- Jaakkola P, Mole DR, Tian YM, et al.: Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292 (5516): 468-72, 2001.
- Keith B, Johnson RS, Simon MC: HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 12 (1): 9-22, 2012.
- Gordan JD, Bertout JA, Hu CJ, et al.: HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 11 (4): 335-47, 2007.
- Koh MY, Lemos R, Liu X, et al.: The hypoxia-associated factor switches cells from HIF-1α- to HIF-2α-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res 71 (11): 4015-27, 2011.
- Koh MY, Darnay BG, Powis G: Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and ubiquitinates hypoxia-inducible factor 1alpha, leading to its oxygen-independent degradation. Mol Cell Biol 28 (23): 7081-95, 2008.
- Monzon FA, Alvarez K, Peterson L, et al.: Chromosome 14q loss defines a molecular subtype of clear-cell renal cell carcinoma associated with poor prognosis. Mod Pathol 24 (11): 1470-9, 2011.
- Kondo K, Klco J, Nakamura E, et al.: Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 1 (3): 237-46, 2002.
- Kondo K, Kim WY, Lechpammer M, et al.: Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol 1 (3): E83, 2003.
- Zimmer M, Doucette D, Siddiqui N, et al.: Inhibition of hypoxia-inducible factor is sufficient for growth suppression of VHL-/- tumors. Mol Cancer Res 2 (2): 89-95, 2004.
- Zimmer M, Ebert BL, Neil C, et al.: Small-molecule inhibitors of HIF-2a translation link its 5'UTR iron-responsive element to oxygen sensing. Mol Cell 32 (6): 838-48, 2008.
- Metelo AM, Noonan HR, Li X, et al.: Pharmacological HIF2α inhibition improves VHL disease-associated phenotypes in zebrafish model. J Clin Invest 125 (5): 1987-97, 2015.
- Scheuermann TH, Li Q, Ma HW, et al.: Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat Chem Biol 9 (4): 271-6, 2013.
- Pan J, Snell W: The primary cilium: keeper of the key to cell division. Cell 129 (7): 1255-7, 2007.
- Simons M, Walz G: Polycystic kidney disease: cell division without a c(l)ue? Kidney Int 70 (5): 854-64, 2006.
- Thoma CR, Frew IJ, Hoerner CR, et al.: pVHL and GSK3beta are components of a primary cilium-maintenance signalling network. Nat Cell Biol 9 (5): 588-95, 2007.
- Hergovich A, Lisztwan J, Barry R, et al.: Regulation of microtubule stability by the von Hippel-Lindau tumour suppressor protein pVHL. Nat Cell Biol 5 (1): 64-70, 2003.
- Hergovich A, Lisztwan J, Thoma CR, et al.: Priming-dependent phosphorylation and regulation of the tumor suppressor pVHL by glycogen synthase kinase 3. Mol Cell Biol 26 (15): 5784-96, 2006.
- Roe JS, Kim HR, Hwang IY, et al.: von Hippel-Lindau protein promotes Skp2 destabilization on DNA damage. Oncogene 30 (28): 3127-38, 2011.
- Kim J, Jonasch E, Alexander A, et al.: Cytoplasmic sequestration of p27 via AKT phosphorylation in renal cell carcinoma. Clin Cancer Res 15 (1): 81-90, 2009.
- Roe JS, Youn HD: The positive regulation of p53 by the tumor suppressor VHL. Cell Cycle 5 (18): 2054-6, 2006.
- Roe JS, Kim H, Lee SM, et al.: p53 stabilization and transactivation by a von Hippel-Lindau protein. Mol Cell 22 (3): 395-405, 2006.
- Ohh M, Yauch RL, Lonergan KM, et al.: The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell 1 (7): 959-68, 1998.
- Lolkema MP, Gervais ML, Snijckers CM, et al.: Tumor suppression by the von Hippel-Lindau protein requires phosphorylation of the acidic domain. J Biol Chem 280 (23): 22205-11, 2005.
- Hall DP, Cost NG, Hegde S, et al.: TRPM3 and miR-204 establish a regulatory circuit that controls oncogenic autophagy in clear cell renal cell carcinoma. Cancer Cell 26 (5): 738-53, 2014.
- Mikhaylova O, Stratton Y, Hall D, et al.: VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell 21 (4): 532-46, 2012.
- Haase VH, Glickman JN, Socolovsky M, et al.: Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci U S A 98 (4): 1583-8, 2001.
- Frew IJ, Thoma CR, Georgiev S, et al.: pVHL and PTEN tumour suppressor proteins cooperatively suppress kidney cyst formation. EMBO J 27 (12): 1747-57, 2008.
- Hickey MM, Lam JC, Bezman NA, et al.: von Hippel-Lindau mutation in mice recapitulates Chuvash polycythemia via hypoxia-inducible factor-2alpha signaling and splenic erythropoiesis. J Clin Invest 117 (12): 3879-89, 2007.
- Varela I, Tarpey P, Raine K, et al.: Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469 (7331): 539-42, 2011.
- Dalgliesh GL, Furge K, Greenman C, et al.: Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 463 (7279): 360-3, 2010.
- Peña-Llopis S, Vega-Rubín-de-Celis S, Liao A, et al.: BAP1 loss defines a new class of renal cell carcinoma. Nat Genet 44 (7): 751-9, 2012.
Clinical Manifestations
Age Ranges and Cumulative Risk of Different Syndrome-Related Neoplasms
The age of onset for von Hippel-Lindau disease (VHL) varies both between different families and between members of the same family. This fact informs the guidelines for starting age and frequency of presymptomatic surveillance examinations. Of all VHL manifestations, retinal hemangioblastomas and pheochromocytomas (PHEOs) have the youngest age of onset; hence, targeted screening is recommended in children younger than 10 years. At least one study has demonstrated that the incidence of new lesions varies depending on patient age, the underlying pathogenic variant, and the organ involved. Examples of reported mean ages and age ranges of VHL clinical manifestations are summarized in Table 1.
| Neoplasm | Mean Age (Range) in y | Cumulative Risk (%) |
|---|---|---|
| PHEO = pheochromocytoma | ||
| aAdapted from Choyke et al. and Lonser et al. | ||
| bLimited data are available for cystadenomas of the broad/round ligament and epididymis. | ||
| Renal cell carcinoma | 37 (16–67) | 24–45 |
| PHEO | 30 (5–58) | 10–20 |
| Pancreatic tumor or cyst | 36 (5–70) | 35–70 |
| Retinal hemangioblastoma | 25 (1–67) | 25–60 |
| Cerebellar hemangioblastoma | 33 (9–78) | 44–72 |
| Brainstem hemangioblastoma | 32 (12–46) | 10–25 |
| Spinal cord hemangioblastoma | 33 (12–66) | 13–50 |
| Endolymphatic sac tumor | 22 (12–50) | 10 |
For more information, see the Clinical Diagnosis of VHL section.
References
- Binderup ML, Budtz-Jørgensen E, Bisgaard ML: Risk of new tumors in von Hippel-Lindau patients depends on age and genotype. Genet Med 18 (1): 89-97, 2016.
- Choyke PL, Glenn GM, Walther MM, et al.: von Hippel-Lindau disease: genetic, clinical, and imaging features. Radiology 194 (3): 629-42, 1995.
- Lonser RR, Glenn GM, Walther M, et al.: von Hippel-Lindau disease. Lancet 361 (9374): 2059-67, 2003.
Tissue Manifestations
Renal Manifestations
More than 55% of individuals with von Hippel-Lindau disease (VHL) only develop multiple renal cell cysts. VHL-associated renal cell carcinomas (RCCs) are characteristically multifocal and bilateral. These RCCs present as masses with both cystic and solid characteristics. In VHL, the cumulative risk of RCC was reported to be 24% to 45%. RCCs smaller than 3 cm tend to be low-grade (Fuhrman nuclear grade 2) and minimally invasive. However, their growth rate varies widely. An investigation of 228 renal lesions in 28 patients who were followed for at least 1 year showed that transition from a simple cyst to a solid lesion was infrequent. Complex cystic and solid lesions contained neoplastic tissue that was uniformly enlarged. These data may be used to predict the progression of renal lesions (from benign to malignant) in patients with VHL. Figure 1 depicts bilateral renal tumors in a patient with VHL.
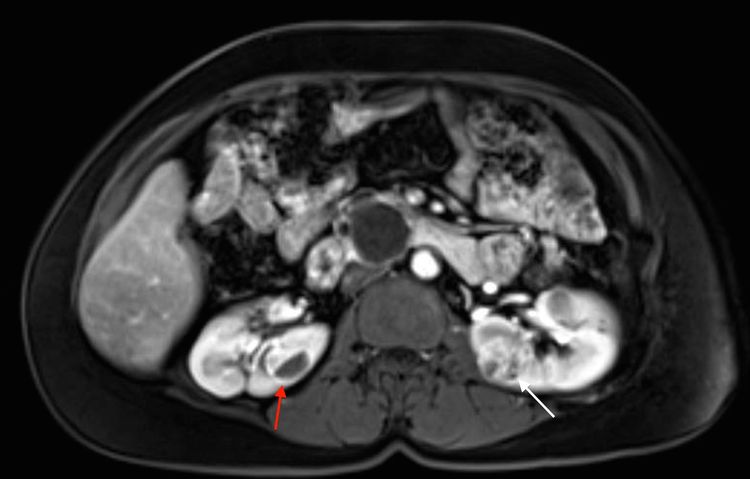 Figure 1. von Hippel-Lindau disease–associated renal cell cancers are characteristically multifocal and bilateral and present as combined cystic and solid masses. The red arrow shows a lesion with a solid and cystic component, and the white arrow shows a predominantly solid lesion.
Figure 1. von Hippel-Lindau disease–associated renal cell cancers are characteristically multifocal and bilateral and present as combined cystic and solid masses. The red arrow shows a lesion with a solid and cystic component, and the white arrow shows a predominantly solid lesion.
Tumors larger than 3 cm may increase in grade as they grow, and metastasis may occur. RCCs often remain asymptomatic for long periods of time.
Retinal Hemangioblastomas
Retinal manifestations, which were first reported more than a century ago, were one of the first recognized VHL features. Retinal hemangioblastomas (also known as capillary retinal angiomas) are one of the most common manifestations of VHL and are present in more than 50% of patients. Retinal involvement is one of the earliest manifestations of VHL, with a mean age of onset at 25 years. These tumors are the first VHL manifestation in nearly 80% of affected individuals and may occur in children as young as 1 year. Sporadic retinal hemangioblastomas are rare; one registry study reported that 84% of these tumors were associated with VHL.
Retinal hemangioblastomas occur most frequently in the periphery of the retina. They can also occur in other locations like the optic nerve, which is a more difficult area to treat. Retinal hemangioblastomas are bright orange spherical tumors that are supplied by a tortuous vascular supply. Nearly 50% of patients have bilateral retinal hemangioblastomas. The median number of lesions per affected eye is six. Patients with VHL can also have other retinal lesions, including retinal vascular hamartomas and flat vascular tumors located in the superficial aspect of the retina.
Longitudinal studies help explain the natural history of these tumors. If left untreated, retinal hemangioblastomas can be a major source of morbidity in patients with VHL. Approximately 8% of patients will develop blindness caused by various mechanisms like secondary maculopathy; this can contribute to retinal detachment or directly cause retinal neurodegeneration. One study suggested that visual impairment severity (including the need for enucleation and the development of phthisis) may depend on a patient's VHL genotype. Patients with symptomatic retinal lesions often have many large retinal hemangioblastomas. Long-term follow-up studies demonstrate that most retinal lesions grow slowly and that new lesions do not develop frequently.
Cerebellar and Spinal Hemangioblastomas
Hemangioblastomas are the most common disease manifestation in patients with VHL, affecting more than 70% of individuals. A prospective study assessed the natural history of hemangioblastomas. The mean age at onset of central nervous system (CNS) hemangioblastomas is 29.1 years (range, 7–73 y). CNS hemangioblastomas were most commonly seen in the cerebellum (45%), spinal cord (36%), cauda equina (11%), and brain stem (7%). While sporadic hemangioblastomas are generally solitary in nature, the VHL-associated CNS lesions are often multifocal. After a mean follow-up of 7 years, 72% of the 225 patients studied developed new lesions. Figures 2 and 3 depict cerebellar and spinal hemangioblastomas, respectively, in patients with VHL.
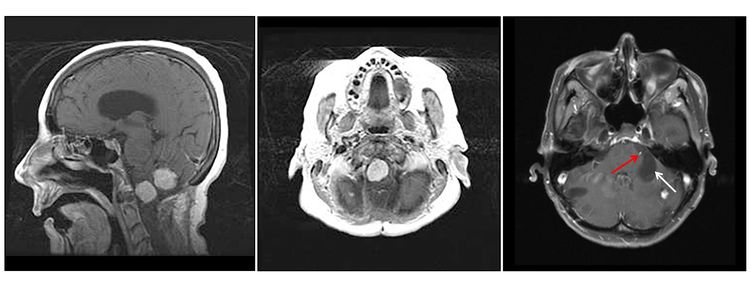 Figure 2. Hemangioblastomas are the most common disease manifestation in patients with von Hippel-Lindau disease. The left panel shows a sagittal view of brainstem and cerebellar lesions. The middle panel shows an axial view of a brainstem lesion. The right panel shows a cerebellar lesion (red arrow) with a dominant cystic component (white arrow).
Figure 2. Hemangioblastomas are the most common disease manifestation in patients with von Hippel-Lindau disease. The left panel shows a sagittal view of brainstem and cerebellar lesions. The middle panel shows an axial view of a brainstem lesion. The right panel shows a cerebellar lesion (red arrow) with a dominant cystic component (white arrow).
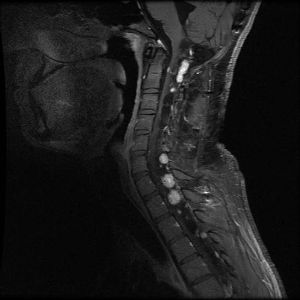 Figure 3. Hemangioblastomas are the most common disease manifestation in patients with von Hippel-Lindau disease. Multiple spinal cord hemangioblastomas are shown.
Figure 3. Hemangioblastomas are the most common disease manifestation in patients with von Hippel-Lindau disease. Multiple spinal cord hemangioblastomas are shown.
Pheochromocytomas and Paragangliomas
The rate of pheochromocytoma (PHEO) formation in the VHL patient population is 25% to 30%. Of patients with VHL-associated PHEOs, 44% developed disease in both adrenal glands. The rate of malignant transformation is very low. Levels of plasma and urine normetanephrine are typically elevated in patients with VHL, and approximately two-thirds will experience physical manifestations such as hypertension, tachycardia, and palpitations. Patients with a partial loss of VHL function (type 2 disease) are at a higher risk of developing PHEOs than VHL patients with a complete loss of VHL function (type 1 disease); the latter develop PHEOs very rarely. In a cohort of 182 patients with PHEOs and paragangliomas (PGLs) that were not associated with a VHL diagnosis, only one patient had a VHLgermlinepathogenic variant.
PGLs are rare in VHL patients but can occur in the head and neck or in the abdomen. A review of VHL patients who developed PHEOs and/or PGLs revealed that 90% of patients manifested PHEOs and only 19% presented with a PGL.
The mean age at diagnosis of VHL-related PHEOs and PGLs is approximately 30 years. Patients with multiple tumors were diagnosed more than a decade earlier than patients with solitary lesions in one series (19 vs. 34 y; P< .001). PHEO diagnosis occurred in patients as young as age 5 years in one cohort, providing a rationale for early testing. All 21 pediatric patients with PHEOs in this 273-patient cohort had elevated plasma normetanephrines.
Pancreatic Manifestations
Patients with VHL may develop multiple serous cystadenomas, pancreatic neuroendocrine tumors (NETs), and simple pancreatic cysts. One study reported that 15% of patients with VHL developed pancreatic NETs. VHL patients do not have an increased risk of pancreatic adenocarcinoma. Serous cystadenomas are benign tumors and warrant no intervention. VHL patients can develop many pancreatic cysts, but these cysts rarely cause symptomatic biliary duct obstruction. Endocrine function is nearly always maintained; however, patients with extensive cystic disease may require pancreatic surgery and pancreatic exocrine supplementation.
Pancreatic cysts and cystadenomas are not malignant, but pancreatic NETs possess malignant characteristics. Pancreatic NETs are usually nonfunctional, but they can metastasize to the lymph nodes and the liver. The risk of pancreatic NET metastasis was analyzed in a large cohort of patients, in which the mean age of pancreatic NET diagnosis was 38 years (range, 16–68 y). The risk of metastasis was lower in patients with small primary lesions (≤3 cm), in patients without an exon 3 pathogenic variant, and in patients whose tumor had a slow doubling time (>500 days). Nonfunctional pancreatic NETs can be monitored by imaging surveillance with intervention when tumors reach 3 cm. Lesions in the head of the pancreas can be considered for surgery at a smaller size to limit operative complexity.
Endolymphatic Sac Tumors (ELSTs)
ELSTs are adenomatous tumors arising from the endolymphatic duct or sac within the posterior part of the petrous bone. ELSTs are rare in the sporadic setting, but are apparent on imaging in 11% to 16% of patients with VHL. Although these tumors do not metastasize, they are locally invasive, eroding through the petrous bone and the inner ear structures. Approximately 30% of VHL patients with ELSTs have bilateral lesions.
ELSTs are an important cause of morbidity in VHL patients. ELSTs evident on imaging are associated with a variety of symptoms, including hearing loss (95% of patients), tinnitus (92%), vestibular symptoms (such as vertigo or disequilibrium) (62%), aural fullness (29%), and facial paresis (8%). In approximately half of patients, symptoms (particularly hearing loss) can occur suddenly, probably as a result of acute intralabyrinthine hemorrhage. Hearing loss or vestibular dysfunction in VHL patients can also present in the absence of radiologically evident ELSTs (approximately 60% of all symptomatic patients) and is believed to be a consequence of microscopic ELSTs.
Hearing loss related to ELSTs is typically irreversible; serial imaging to enable early detection of ELSTs in asymptomatic patients and resection of radiologically evident lesions are important components in the management of VHL patients. Surgical resection by retrolabyrinthine posterior petrosectomy is usually curative and can prevent onset or worsening of hearing loss; this procedure can also improve vestibular symptoms.
Broad/Round Ligament Papillary Cystadenomas
Tumors of the broad ligament can occur in females with VHL and are known as papillary cystadenomas. These tumors are extremely rare, and fewer than 20 have been reported in the literature. Papillary cystadenomas are histologically identical to epididymal cystadenomas, which are commonly observed in males with VHL. One important difference is that papillary cystadenomas are almost exclusively observed in patients with VHL, whereas epididymal cystadenomas can occur sporadically. These tumors are frequently cystic, and although they can become large, they generally have fairly indolent behavior.
Epididymal Cystadenomas
Fluid-filled epididymal cysts, or spermatoceles, are very common in adult men. In VHL, the epididymis can contain more complex cystic neoplasms known as papillary cystadenomas, which are rare in the general population. More than one-third of all cases of epididymal cystadenomas reported in the literature and most cases of bilateral cystadenomas have been reported in patients with VHL. These well-circumscribed lesions have variable amounts of cystic and papillary components that are lined with epithelial cuboidal or columnar clear cells. Among symptomatic patients, the most common presentation of epididymal cystadenoma is a painless, slow-growing scrotal swelling. The differential diagnoses of epididymal tumors include adenomatoid tumor (which is the most common tumor in this site), metastatic ccRCC, and papillary mesothelioma.
In a small series, histological analysis did not reveal features typically associated with malignancy, such as mitotic figures, nuclear pleomorphism, and necrosis. Lesions were strongly positive for CK7 and negative for RCC. Carbonic anhydrase IX (CAIX) was positive in all tumors. PAX8 was positive in most cases. These features were reminiscent of clear cell papillary RCC, a relatively benign form of RCC without known metastatic potential.
References
- Choyke PL, Glenn GM, Walther MM, et al.: The natural history of renal lesions in von Hippel-Lindau disease: a serial CT study in 28 patients. AJR Am J Roentgenol 159 (6): 1229-34, 1992.
- Poston CD, Jaffe GS, Lubensky IA, et al.: Characterization of the renal pathology of a familial form of renal cell carcinoma associated with von Hippel-Lindau disease: clinical and molecular genetic implications. J Urol 153 (1): 22-6, 1995.
- Walther MM, Choyke PL, Glenn G, et al.: Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol 161 (5): 1475-9, 1999.
- Walther MM, Lubensky IA, Venzon D, et al.: Prevalence of microscopic lesions in grossly normal renal parenchyma from patients with von Hippel-Lindau disease, sporadic renal cell carcinoma and no renal disease: clinical implications. J Urol 154 (6): 2010-4; discussion 2014-5, 1995.
- Chew EY: Ocular manifestations of von Hippel-Lindau disease: clinical and genetic investigations. Trans Am Ophthalmol Soc 103: 495-511, 2005.
- Choyke PL, Glenn GM, Walther MM, et al.: von Hippel-Lindau disease: genetic, clinical, and imaging features. Radiology 194 (3): 629-42, 1995.
- Lonser RR, Glenn GM, Walther M, et al.: von Hippel-Lindau disease. Lancet 361 (9374): 2059-67, 2003.
- Dollfus H, Massin P, Taupin P, et al.: Retinal hemangioblastoma in von Hippel-Lindau disease: a clinical and molecular study. Invest Ophthalmol Vis Sci 43 (9): 3067-74, 2002.
- Wong WT, Agrón E, Coleman HR, et al.: Clinical characterization of retinal capillary hemangioblastomas in a large population of patients with von Hippel-Lindau disease. Ophthalmology 115 (1): 181-8, 2008.
- Binderup MLM, Stendell AS, Galanakis M, et al.: Retinal hemangioblastoma: prevalence, incidence and frequency of underlying von Hippel-Lindau disease. Br J Ophthalmol 102 (7): 942-947, 2018.
- Kreusel KM, Bechrakis NE, Krause L, et al.: Retinal angiomatosis in von Hippel-Lindau disease: a longitudinal ophthalmologic study. Ophthalmology 113 (8): 1418-24, 2006.
- Schmidt D, Neumann HP: Retinal vascular hamartoma in von Hippel-Lindau disease. Arch Ophthalmol 113 (9): 1163-7, 1995.
- Wittström E, Nordling M, Andréasson S: Genotype-phenotype correlations, and retinal function and structure in von Hippel-Lindau disease. Ophthalmic Genet 35 (2): 91-106, 2014.
- Reich M, Jaegle S, Neumann-Haefelin E, et al.: Genotype-phenotype correlation in von Hippel-Lindau disease. Acta Ophthalmol 99 (8): e1492-e1500, 2021.
- Toy BC, Agrón E, Nigam D, et al.: Longitudinal analysis of retinal hemangioblastomatosis and visual function in ocular von Hippel-Lindau disease. Ophthalmology 119 (12): 2622-30, 2012.
- Huntoon K, Wu T, Elder JB, et al.: Biological and clinical impact of hemangioblastoma-associated peritumoral cysts in von Hippel-Lindau disease. J Neurosurg 124 (4): 971-6, 2016.
- Kanno H, Kuratsu J, Nishikawa R, et al.: Clinical features of patients bearing central nervous system hemangioblastoma in von Hippel-Lindau disease. Acta Neurochir (Wien) 155 (1): 1-7, 2013.
- Lonser RR, Butman JA, Huntoon K, et al.: Prospective natural history study of central nervous system hemangioblastomas in von Hippel-Lindau disease. J Neurosurg 120 (5): 1055-62, 2014.
- Walther MM, Reiter R, Keiser HR, et al.: Clinical and genetic characterization of pheochromocytoma in von Hippel-Lindau families: comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma. J Urol 162 (3 Pt 1): 659-64, 1999.
- Aufforth RD, Ramakant P, Sadowski SM, et al.: Pheochromocytoma Screening Initiation and Frequency in von Hippel-Lindau Syndrome. J Clin Endocrinol Metab 100 (12): 4498-504, 2015.
- Welander J, Söderkvist P, Gimm O: Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer 18 (6): R253-76, 2011.
- Eisenhofer G, Walther MM, Huynh TT, et al.: Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab 86 (5): 1999-2008, 2001.
- Zbar B, Kishida T, Chen F, et al.: Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat 8 (4): 348-57, 1996.
- Chen F, Slife L, Kishida T, et al.: Genotype-phenotype correlation in von Hippel-Lindau disease: identification of a mutation associated with VHL type 2A. J Med Genet 33 (8): 716-7, 1996.
- Friedrich CA: Genotype-phenotype correlation in von Hippel-Lindau syndrome. Hum Mol Genet 10 (7): 763-7, 2001.
- Eisenhofer G, Vocke CD, Elkahloun A, et al.: Genetic screening for von Hippel-Lindau gene mutations in non-syndromic pheochromocytoma: low prevalence and false-positives or misdiagnosis indicate a need for caution. Horm Metab Res 44 (5): 343-8, 2012.
- Shuch B, Ricketts CJ, Metwalli AR, et al.: The genetic basis of pheochromocytoma and paraganglioma: implications for management. Urology 83 (6): 1225-32, 2014.
- Eisenhofer G, Timmers HJ, Lenders JW, et al.: Age at diagnosis of pheochromocytoma differs according to catecholamine phenotype and tumor location. J Clin Endocrinol Metab 96 (2): 375-84, 2011.
- Charlesworth M, Verbeke CS, Falk GA, et al.: Pancreatic lesions in von Hippel-Lindau disease? A systematic review and meta-synthesis of the literature. J Gastrointest Surg 16 (7): 1422-8, 2012.
- Libutti SK, Choyke PL, Bartlett DL, et al.: Pancreatic neuroendocrine tumors associated with von Hippel Lindau disease: diagnostic and management recommendations. Surgery 124 (6): 1153-9, 1998.
- Blansfield JA, Choyke L, Morita SY, et al.: Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs). Surgery 142 (6): 814-8; discussion 818.e1-2, 2007.
- Manski TJ, Heffner DK, Glenn GM, et al.: Endolymphatic sac tumors. A source of morbid hearing loss in von Hippel-Lindau disease. JAMA 277 (18): 1461-6, 1997.
- Choo D, Shotland L, Mastroianni M, et al.: Endolymphatic sac tumors in von Hippel-Lindau disease. J Neurosurg 100 (3): 480-7, 2004.
- Megerian CA, Haynes DS, Poe DS, et al.: Hearing preservation surgery for small endolymphatic sac tumors in patients with von Hippel-Lindau syndrome. Otol Neurotol 23 (3): 378-87, 2002.
- Kim HJ, Butman JA, Brewer C, et al.: Tumors of the endolymphatic sac in patients with von Hippel-Lindau disease: implications for their natural history, diagnosis, and treatment. J Neurosurg 102 (3): 503-12, 2005.
- Lonser RR, Kim HJ, Butman JA, et al.: Tumors of the endolymphatic sac in von Hippel-Lindau disease. N Engl J Med 350 (24): 2481-6, 2004.
- Nogales FF, Goyenaga P, Preda O, et al.: An analysis of five clear cell papillary cystadenomas of mesosalpinx and broad ligament: four associated with von Hippel-Lindau disease and one aggressive sporadic type. Histopathology 60 (5): 748-57, 2012.
- Cox R, Vang R, Epstein JI: Papillary cystadenoma of the epididymis and broad ligament: morphologic and immunohistochemical overlap with clear cell papillary renal cell carcinoma. Am J Surg Pathol 38 (5): 713-8, 2014.
- Brady A, Nayar A, Cross P, et al.: A detailed immunohistochemical analysis of 2 cases of papillary cystadenoma of the broad ligament: an extremely rare neoplasm characteristic of patients with von hippel-lindau disease. Int J Gynecol Pathol 31 (2): 133-40, 2012.
- Odrzywolski KJ, Mukhopadhyay S: Papillary cystadenoma of the epididymis. Arch Pathol Lab Med 134 (4): 630-3, 2010.
- Uppuluri S, Bhatt S, Tang P, et al.: Clear cell papillary cystadenoma with sonographic and histopathologic correlation. J Ultrasound Med 25 (11): 1451-3, 2006.
- Vijayvargiya M, Jain D, Mathur SR, et al.: Papillary cystadenoma of the epididymis associated with von Hippel-Lindau disease diagnosed on fine needle aspiration cytology. Cytopathology 25 (4): 279-81, 2014.
Genetic Risk Assessment for von Hippel-Lindau Disease (VHL)
The primary risk factor for VHL (or any hereditary forms of renal cancer) is an affected family member. Risk assessment should also consider gender and age for specific VHL-related neoplasms. For example, pheochromocytomas (PHEOs) may develop in early childhood, sometimes occurring in patients as young as 8 years. Gender-specific VHL clinical findings include epididymal cystadenomas in males (10%–26%), which are virtually pathognomonic for VHL, especially when bilateral. These lesions occur rarely in the general population. Epididymal cysts are also common in VHL, but they are reported in 23% of the general population, making them a poor diagnostic discriminator. Females have lesions that are histologically similar to cystadenomas, which occur in the broad ligament.
Each child of an individual with VHL has a 50% chance of inheriting the VHLvariantallele from the affected parent.
Clinical Diagnosis of VHL
Diagnosis of VHL is frequently based on clinical criteria. If there is family history of VHL, a previously unevaluated family member can be clinically diagnosed with VHL if this person presents with one or more VHL-related tumors (e.g., CNS/retinal hemangioblastomas, PHEOs, ccRCCs, or endolymphatic sac tumors). If a patient does not have a family history of VHL, he/she must meet one of the following criteria: (1) two or more CNS hemangioblastomas, or (2) one CNS hemangioblastoma and either (a) a visceral tumor or (b) an endolymphatic sac tumor. For more information about VHL diagnostic details, see Table 2.
In 1998, all germlinepathogenic variants identified in a cohort of 93 VHL families were reported. Since then, VHL diagnosis has been based on a combination of the following: (1) identifying clinical VHL-associated manifestations, and (2) conducting genetic testing for VHL pathogenic variants identified within families. This diagnostic strategy can differ for individual family members. Table 2 summarizes a combined approach that uses both methods mentioned above (i.e., identifying VHL clinical manifestations and VHL genetic testing).
| Family History of VHL | Genetic Testing | Scenarios for Clinical Diagnosis | Requirements for Clinical Diagnosis |
|---|---|---|---|
| CNS = central nervous system; ccRCC = clear cell renal cell carcinoma; PHEO = pheochromocytoma. | |||
| Adapted and updated from Glenn et al. and Pithukpakorn and Glenn. | |||
| With a family history of VHL | Test DNA for the same VHL pathogenic variant as previously identified in an affected biological relative(s) | When the VHL pathogenic variant in a biological relative is unknown | At least one of the following is required for clinical diagnosis: |
| - Epididymal or broad ligament cystadenoma | |||
| - CNS hemangioblastoma | |||
| - Multifocal ccRCC | |||
| - PHEO | |||
| - Retinal hemangioblastoma | |||
| - Pancreatic neuroendocrine tumor | |||
| - Pancreatic cysts and/or cystadenoma | |||
| - Endolymphatic sac tumor | |||
| Without a family history of VHL | Genetic test results may be negative if the VHL pathogenic variant occurred postzygotically (e.g., VHL mosaicism) | When the VHL pathogenic variant is unknown or germline negative, but there are clinical signs of VHL | At least one of the following is required for clinical diagnosis: |
| - CNS hemangioblastoma | |||
| - Retinal hemangioblastoma | |||
| If only one of the above is present, then one of the following is also required for a clinical diagnosis: | |||
| - ccRCC | |||
| - PHEO | |||
| - Pancreatic cysts and/or cystadenoma | |||
| - Endolymphatic sac tumor | |||
| - Epididymal or broad ligament cystadenoma | |||
Genetic Testing in VHL
It is recommended that at-risk family members be informed that genetic testing for VHL is available. A family member with a clinical diagnosis of VHL, or one who is showing signs/symptoms of VHL, is generally offered genetic testing first. Germline pathogenic variants in VHL are detected in more than 99% of families affected by VHL. Sequence analysis of all three exons detects point variants in the VHLgene (~72% of all pathogenic variants). Deletions are detected mainly by using next-generation sequencing (NGS), with confirmation using targeted chromosomal microarray and/or multiplex ligation-dependent probe amplification. Array comparative genomic hybridization is also used to identify genomic imbalances. Anecdotal evidence suggests that NGS may be used for suspected mosaicism cases after receipt of a negative VHL genetic test.
Genetic counseling is provided before genetic testing. Such counseling includes a discussion of the medical, economic, and psychosocial implications for the patient and his/her blood relatives. After genetic counseling occurs, the patient may choose to proceed with genetic testing, after providing informed consent. Additional genetic counseling is given when results are reported to the patient. When a VHL pathogenic variant is identified in a family, biological relatives who test negative for this variant are not carriers of the trait (i.e., they are true negatives) and are not predisposed to VHL manifestations. Moreover, the children of true-negative family members are also not at risk of developing VHL. Clinical testing throughout their lifetimes is, therefore, unnecessary.
Genetic Diagnosis in VHL
A germline pathogenic variant in the VHL gene is considered a genetic diagnosis.
This finding predisposes an individual to clinical VHL and confers a 50% risk for offspring to inherit the VHL pathogenic variant. Approximately 400 unique pathogenic variants in the VHL gene have been associated with clinical VHL, and their presence verifies the disease-causing capability of the variant. The diagnostic genetic evaluation in a previously untested family generally begins with a clinically diagnosed individual. If a VHL pathogenic variant is identified, that specific pathogenic variant becomes the DNA marker for which other biological relatives are tested. Some individuals may meet VHL clinical criteria for diagnosis but do not test positive for a VHL pathogenic variant. When these individuals also do not have family history of VHL, a de novo pathogenic variant or mosaicism may be present. The latter may be detected by performing genetic testing on other bodily tissues, such as skin fibroblasts or exfoliated buccal cells. For more information, see the De novo pathogenic variants and mosaicism section.
References
- Choyke PL, Glenn GM, Walther MM, et al.: von Hippel-Lindau disease: genetic, clinical, and imaging features. Radiology 194 (3): 629-42, 1995.
- Maher ER, Yates JR, Harries R, et al.: Clinical features and natural history of von Hippel-Lindau disease. Q J Med 77 (283): 1151-63, 1990.
- Lonser RR, Glenn GM, Walther M, et al.: von Hippel-Lindau disease. Lancet 361 (9374): 2059-67, 2003.
- Pithukpakorn M, Glenn G: von Hippel-Lindau syndrome. Community Oncology 1 (4): 232-43, 2004.
- Glenn GM, Daniel LN, Choyke P, et al.: Von Hippel-Lindau (VHL) disease: distinct phenotypes suggest more than one mutant allele at the VHL locus. Hum Genet 87 (2): 207-10, 1991.
- Stolle C, Glenn G, Zbar B, et al.: Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat 12 (6): 417-23, 1998.
- Coppin L, Grutzmacher C, Crépin M, et al.: VHL mosaicism can be detected by clinical next-generation sequencing and is not restricted to patients with a mild phenotype. Eur J Hum Genet 22 (9): 1149-52, 2014.
Screening for Early Detection of VHL Manifestations
Screening guidelines have been suggested for various manifestations of VHL. In general, these recommendations are based on expert opinion and consensus, but most of these recommendations are not evidence-based. Several VHL screening and surveillance guidelines are available. The VHL Alliance's Suggested Active Surveillance Guidelines is an example of a surveillance plan that is tailored for each individual with VHL. These guidelines are based on a consensus from a panel of experts who work in various disciplines. The modalities in the VHL Alliance's surveillance plan may be used to guide initial clinical diagnostic testing of individuals with suspected VHL (see Table 3). These modalities may also be used for periodic surveillance of at-risk individuals to detect VHL-associated neoplasms at an early stage. Periodic, presymptomatic screening is advised for at-risk individuals, including those who tested positive for a VHL pathogenic variant and those who chose not to be tested for a VHL pathogenic variant but have biological relatives with VHL. The risk of inheriting a VHL pathogenic variant in these individuals may be as high as 50%.
| Examination/Test | Condition Screened For |
|---|---|
| CNS = central nervous system; IAC = internal auditory canal; MRI = magnetic resonance imaging. | |
| aAdapted from VHL Alliance. | |
| bAge-appropriate history and physical examination includes the following: neurological examination, auditory/vestibuloneural questioning and testing, visual symptoms, catecholamine-excess symptom assessment (headaches, palpitations, diaphoresis, hyperactivity, anxiety, polyuria, and abdominal pain). | |
| History and physical examinationb | All conditions listed in this table |
| Dilated, in-person eye examination with ophthalmoscopy | Retinal hemangioblastoma |
| Blood pressure and pulse measurements, plasma free metanephrines or fractionated 24-hour urinary free metanephrines test | Pheochromocytoma/paraganglioma |
| MRI of brain and total spine with and without contrast | CNS hemangioblastoma |
| MRI of abdomen with and without contrast | Renal cell carcinoma, pheochromocytoma/paraganglioma, pancreatic neuroendocrine tumor/cyst |
| Audiological exam, MRI of IAC | Endolymphatic sac tumor |
Level of evidence: 5
Screening for PHEOs
PHEOs can be detected early when key tests like catecholamine/metanephrine levels and cross-sectional abdominal imaging are done. Most small (≤1 cm) PHEOs can have undetectable levels of catecholamines/metanephrines, and thus, these levels can increase with PHEO tumor progression.
Biochemical testing for PHEOs
Biochemical testing is critical when evaluating individuals with VHL, since metabolite levels can often be elevated in the absence of anatomic imaging findings. Assessment begins in childhood, with some guidelines recommending initiation at age 5 years (Table 3). Clinicians have the option of performing plasma testing, urinary testing, or both. Catecholamines levels can vary greatly due to diet and medication use. However, measurement of their metabolites, like metanephrines, is suggested because this has higher performance metrics than that of catecholamines. A fourfold or greater elevation of metanephrines suggests that a PHEO or paraganglioma (PGL) may be present. Individuals with VHL-associated PHEOs often have isolated normetanephrines, whereas PHEOs in other endocrine syndromes show a different functional profile. For more information about biochemical testing for PHEOs, see the Clinical Diagnosis of PGL and PHEO section in Genetics of Endocrine and Neuroendocrine Neoplasias.
Imaging for PHEOs
Cross-sectional imaging is initiated early in the second decade of life to evaluate the kidneys, adrenal glands, and pancreas. Both magnetic resonance imaging (MRI) and computed tomography (CT) scans have excellent performance characteristics for detecting PHEOs, with a sensitivity greater than 90%. Additional imaging studies may provide clinical utility when biochemical studies increase clinical suspicion for PHEOs/PGLs but the patient does not have visible lesions. While most tumors that arise from chromaffin tissue in VHL are PHEOs, PGLs can also occur in the chest, abdomen, pelvis, and head/neck. Dedicated cross-sectional imaging can be performed in these areas in addition to whole-body functional imaging. For more information on imaging methods for sporadic and hereditary PHEOs, see the Clinical Diagnosis of PGL and PHEO section in Genetics of Endocrine and Neuroendocrine Neoplasias. Functional imaging studies like scintigraphy (nuclear medicine) or positron emission tomography (PET) scans are useful for locating PHEOs when there is high suspicion and when CT or MRI fails to detect a tumor. Imaging performance can vary based on the tumor's location and genetic background (i.e., the type of hereditary cancer syndrome or genetic variant involved). Iodine I 123 (123I)-metaiodobenzylguanidine scintigraphy coupled with CT imaging provides anatomical and functional information with good sensitivity (80%–90%) and specificity (95%–100%). Other modalities, such as fluorine F 18 (18F)-fluorodopa and 18F-fludeoxyglucose PET/CT, are also useful for tumor localization.
Screening for Endolymphatic Sac Tumors (ELSTs)
ELSTs can often cause permanent audiovestibular dysfunction. Early identification and treatment of these tumors may decrease morbidity significantly. ELST screening typically includes clinical assessment, audiogram, and imaging. The VHL Alliance's expert consensus guideline on screening recommendations for ELSTs is based on available evidence. Since ELSTs in VHL have an average onset of age 30 years (range, 6–62 y), suggested screening begins at age 11 years. A thorough clinical history can identify patients with common audiovestibular symptoms including tinnitus, vertigo, aural fullness, and hearing loss. Annual clinical assessment is also recommended starting at age 11 years, with additional workup if new symptoms are reported. Diagnostic audiograms can identify hearing loss across various frequencies. Audiograms are recommended every 2 years beginning at age 11 years. However, the specificity for ELST identification is low, since hearing loss is common in the general population. Notably though, hearing loss often occurs at younger ages in patients with VHL who have ELSTs. While rare, the identification of ELSTs prior to imaging have been reported. If clinical symptoms of ELSTs occur or the patient has an abnormal audiogram, further assessment via imaging is recommended. MRI is the mainstay test to identify small ELSTs. While an MRI of the brain is useful, a thin-cut MRI scan of the inner auditory canal (IAC) can identify lesions as small as 2 mm in size and has high diagnostic specificity. Intralabyrinthine hemorrhage, hydrops, and soft tissue enhancement are typical findings associated with ELSTs. A baseline MRI of the IAC is suggested between ages 15 to 20 years to assess anatomy. Particular attention to the inner ear is recommended on routine brain imaging as part of hemangioblastoma assessments. A thin-slice CT of the temporal bone is another useful modality for ELST confirmation. While this screening modality can aid in preoperative planning, it does have the inherent risks of radiation. Screening for ELSTs beyond age 65 years has questionable benefit, since there have been no reports of patients with ELSTs beyond age 62 years.
References
- Wolters WPG, Dreijerink KMA, Giles RH, et al.: Multidisciplinary integrated care pathway for von Hippel-Lindau disease. Cancer 128 (15): 2871-2879, 2022.
- Binderup MLM, Smerdel M, Borgwadt L, et al.: von Hippel-Lindau disease: Updated guideline for diagnosis and surveillance. Eur J Med Genet 65 (8): 104538, 2022.
- VHL Alliance: VHLA Suggested Active Surveillance Guidelines. Boston, MA: VHL Alliance, 2020. Available online. Last accessed January 3, 2024.
- Daniels AB, Tirosh A, Huntoon K, et al.: Guidelines for surveillance of patients with von Hippel-Lindau disease: Consensus statement of the International VHL Surveillance Guidelines Consortium and VHL Alliance. Cancer 129 (19): 2927-2940, 2023.
- Sanford T, Gomella PT, Siddiqui R, et al.: Long term outcomes for patients with von Hippel-Lindau and Pheochromocytoma: defining the role of active surveillance. Urol Oncol 39 (2): 134.e1-134.e8, 2021.
- Neary NM, King KS, Pacak K: Drugs and pheochromocytoma--don't be fooled by every elevated metanephrine. N Engl J Med 364 (23): 2268-70, 2011.
- Shuch B, Ricketts CJ, Metwalli AR, et al.: The genetic basis of pheochromocytoma and paraganglioma: implications for management. Urology 83 (6): 1225-32, 2014.
- Čtvrtlík F, Koranda P, Schovánek J, et al.: Current diagnostic imaging of pheochromocytomas and implications for therapeutic strategy. Exp Ther Med 15 (4): 3151-3160, 2018.
- Ilias I, Pacak K: Current approaches and recommended algorithm for the diagnostic localization of pheochromocytoma. J Clin Endocrinol Metab 89 (2): 479-91, 2004.
- Ilias I, Meristoudis G: Functional Imaging of Paragangliomas with an Emphasis on Von Hippel-Lindau-Associated Disease: A Mini Review. J Kidney Cancer VHL 4 (3): 30-36, 2017.
- Mehta GU, Kim HJ, Gidley PW, et al.: Endolymphatic Sac Tumor Screening and Diagnosis in von Hippel-Lindau Disease: A Consensus Statement. J Neurol Surg B Skull Base 83 (Suppl 2): e225-e231, 2022.
- Poulsen ML, Gimsing S, Kosteljanetz M, et al.: von Hippel-Lindau disease: surveillance strategy for endolymphatic sac tumors. Genet Med 13 (12): 1032-41, 2011.
- Butman JA, Nduom E, Kim HJ, et al.: Imaging detection of endolymphatic sac tumor-associated hydrops. J Neurosurg 119 (2): 406-11, 2013.
Management of Disease Manifestations
Management of Renal Tumors
Surgical interventions for renal tumors
The management of von Hippel-Lindau disease (VHL) has changed significantly as clinicians have learned how to balance the risk of cancer dissemination while minimizing renal morbidity. Some initial surgical series performed bilateral radical nephrectomies for renal tumors followed by renal transplantation. Nephron-sparing surgery (NSS) was introduced in the 1980s for VHL after several groups demonstrated that this method conferred a low risk of cancer spread using a less radical surgical approach. In 1995, a large multi-institutional series demonstrated how NSS could produce excellent cancer-specific survival in patients with renal cell carcinoma (RCC). Because of multiple reports of excellent outcomes, NSS is now considered the surgical standard of care for VHL-associated RCCs when technically feasible. Over time, NSS techniques have been refined for patients with VHL to minimize damage to the adjacent normal parenchyma. For example, in traditional NSS, a wide margin of tissue is taken. However, in a subsequent type of NSS called enucleation, much of the adjacent normal parenchyma can be preserved.
Patients with VHL can have dozens of renal tumors. Therefore, resection of all evident renal disease may not be feasible. To minimize the morbidity of multiple surgical procedures, loss of kidney function, and the risk of distant progression, a method to balance over- and under-treatment was sought. The National Cancer Institute (NCI) evaluated a specific tumor-size threshold to trigger surgical intervention. An evaluation of 52 patients with VHL or hereditary papillary renal carcinoma who were treated when their largest solid renal lesion reached 3 cm demonstrated no evidence of distant metastases or the need for renal replacement therapy after a median follow-up period of 60 months. Subsequent retrospective series reinforced the importance of this 3-cm tumor-size threshold. For example, in one study, none of the 108 patients with VHL showed evidence of distant metastases when renal tumors were managed at sizes of 3 cm or smaller. In patients with tumors larger than 3 cm, 27.3% (20 of 73) developed distant recurrences. This 3-cm tumor-size threshold is now widely used in the United States to trigger surgical intervention for VHL-associated clear cell renal cell carcinomas (ccRCCs). However, some international research groups have published data that support active surveillance of renal tumors until they reach 4 cm. When surgery is performed on a patient with VHL, resection of as many renal tumors as is clinically feasible may delay the need for further surgical interventions. The use of intraoperative ultrasound is helpful to identify and remove smaller lesions.
Many patients with VHL develop new RCCs on an ongoing basis and may require further intervention. Adhesions and perinephric scarring make subsequent surgical procedures more challenging. While a radical nephrectomy could be considered, NSS remains the preferred approach, when feasible. While there may be a higher incidence of complications, repeat and salvage NSS can enable patients to maintain excellent renal function and provide promising oncological outcomes at the time of intermediate follow-up. These surgeries may be handled best by specialized centers with significant experience managing individuals with hereditary forms of kidney cancer.
Level of evidence: 3di
Ablative techniques for renal tumors
Thermal ablative techniques apply extreme heat or cold to a mass to destroy it. Cryoablation (CA) and radiofrequency ablation (RFA) were introduced in the late 1990s to manage small renal masses. For sporadic renal masses, both thermal ablative techniques achieved a recurrence-free survival rate of nearly 90%, leading the American Urologic Association to recommend discussing this technique with high-risk patients who have small renal masses (≤4 cm). For patients with VHL, the clinical applications of ablative techniques are still not clearly defined, and surgery remains the most-studied intervention. Ablative techniques were introduced to VHL-associated RCC management in a phase II trial investigating the effects of ablation at the time of lesion resection. In this study, 11 tumors were treated, and an intra-operative ultrasound showed complete elimination of blood flow to the tumors. On final pathology, there was evidence of treatment effect on all tumors. Since that time, some centers have successfully used thermal ablative techniques for primary and salvage management in patients with VHL. Other centers have found that techniques such as RFA have a high failure rate and should be reserved for patients with marginal renal function. Despite limited long-term data, these techniques have been increasingly used to treat RCC in patients with VHL. A single-institution study evaluated treatment trends in RCC in 113 patients with VHL. Between 2004 and 2009, 43% of cases were managed with RFA.
Thermal ablation may play an increasing role as salvage therapy for individuals with a high risk of morbidity from surgery. CA was evaluated as a salvage therapy in 14 patients to avoid the morbidity associated with repeated NSS. Results showed minimal change in renal function after treatment with CA. There was suspicion of recurrence in only 4 of 33 tumors (12.1%) after a median follow-up period of 37 months. However, surgery after thermal ablation is a challenging endeavor, with a significantly higher rate of postoperative complications due to adhesions and scarring, especially along the tract of the ablative probes. Clinicians must consider how thermal ablation could impact future RCC management in younger individuals, who may need further surgical management in their lifetimes.
In summary, the clinical applications of ablative techniques are not clearly defined in VHL, and surgery remains the most-studied intervention. The available clinical evidence suggests that ablative approaches are only recommended for small (≤3 cm), solid-enhancing renal masses in older patients with high operative risk—especially those facing salvage renal surgery because of a high complication rate. Young age, tumors larger than 4 cm, hilar tumors, and cystic lesions are relative contraindications for thermal ablation.
Level of evidence: 3di
Management of Pheochromocytomas (PHEOs)
Surveillance of PHEOs
PHEOs can be a significant source of morbidity in patients with VHL because excess catecholamines can cause significant cardiovascular effects. Many small, asymptomatic PHEOs (≤2 cm) can be safely observed because they have very slow rates of growth and progression. However, because PHEOs in individuals with VHL can become malignant (5%–10% ultimately metastasize), it is imperative that surveillance be performed with very close monitoring to ensure that there is sufficient time to intervene. Since partial adrenalectomy is the best surgical option for smaller PHEOs, the size and anatomical location of PHEOs should be considered when electing surveillance. Special considerations may be needed for patients undergoing future surgeries or childbirth. When a preoperative alpha blockade is not used in patients with a PHEO, these patients can experience a massive release of catecholamines, which can lead to hemodynamic instability.
Surgical interventions for PHEOs
Surgical resection is an important tool for managing PHEOs in individuals with VHL. It is important that all patients have detailed endocrine evaluations and preoperative alpha-blockades prior to surgical resection of PHEOs. Medications are often initiated and carefully titrated prior to surgery to prevent potentially life-threatening cardiovascular complications. For more information, see the Preoperative management section in Genetics of Endocrine and Neuroendocrine Neoplasias.
PHEOs in patients with VHL may be managed differently than in individuals with sporadic PHEOs or other hereditary cancer syndromes. Since PHEOs are multifocal and bilateral in individuals with VHL nearly 50% of the time, many patients have undergone bilateral adrenalectomy and have required lifelong steroid replacement. The morbidity associated with adrenal replacement and the development of Cushing syndrome in individuals with VHL heightened interest in cortical-sparing partial adrenalectomy. Even after extensive adrenal mobilization and tumor resection, the adrenal gland has extensive collateral arterial supply and venous drainage that can permit organ survival. Leaving at least 15% to 30% of residual adrenal gland volume is necessary to allow sufficient hormone production. Most adrenal glands will maintain functional cortisol production after cortical-sparing partial adrenalectomy is performed with modern techniques. In a solitary adrenal gland, a series demonstrated that only 1 of 13 (8%) patients required lifelong steroid replacement.
The possibility of partial adrenalectomy leaving residual cancer behind is a concern in patients with a malignant PHEO. However, in the VHL population, the malignancy rate of PHEOs is low (<5%). The local recurrence rate of PHEOs after partial adrenalectomy also appears to be low (0%–33%). Therefore, when feasible and safe from an oncological perspective, most guidelines recommend partial adrenalectomy for managing PHEOs in patients with VHL.
In a total adrenalectomy, the adrenal vein is usually divided early to limit catecholamine release during gland mobilization. However, in a partial adrenalectomy, dividing the adrenal vein can lead to venous congestion and gland compromise. In a patient with an effective preoperative catecholamine block, it may be possible to only clamp the adrenal vein during the resection and unclamp it after tumor excision. The optimal amount of adjacent normal parenchyma to remove is unclear. Initial surgical approaches for partial adrenalectomy in patients with tumors in the tail/head of the adrenal gland included removing this portion of the gland. However, when patients had tumors in the body of the adrenal gland, this region was resected along with a thin rim of normal parenchyma. As data have clarified the risk of malignancy and local recurrence of PHEOs in patients with VHL, a technique involving enucleative resection of the tumor pseudocapsule has been described (which is similar to renal tumor enucleation techniques). This approach may preserve the maximum amount of cortical tissue and limit vascular compromise to the residual adrenal gland. However, concerns over a higher rate of local recurrence may limit use of this approach.
Both open resection and laparoscopic surgical approaches are safe, but if feasible, laparoscopic removal of adrenal tissue is preferred. Means of exposure and approach are based on the anatomical location of the tumor. Direct access to the adrenal gland and para-aortic region can be achieved with the posterior approach, which is direct, safe, and efficient. Adequate exposure of the complete tumor is important for total removal. Robotic assistance can be used in select cases because it offers a three-dimensional, magnified view of the tumor's anatomy. When an individual has a history of multiple abdominal procedures, a minimally invasive approach is often not feasible because of adhesions. Open resection is commonly recommended for patients with large adrenal tumors because laparoscopy is difficult to perform within a confined space and increases risk of complications. For more information about surgical approaches for PHEOs, see the Surgery section in Genetics of Endocrine and Neuroendocrine Neoplasias.
Management of Pancreatic Manifestations
VHL-related tumors, such as pancreatic neuroendocrine tumors (NETs), may be identified during incidental imaging or lifelong surveillance protocols. The clinical characteristics of the pancreatic lesions (i.e., cystic vs. solid, symptomatic vs. asymptomatic, size) determine whether patients are eligible for conservative management with imaging surveillance or whether they require surgical intervention.
Workup and imaging for pancreatic manifestations
Pancreatic cysts are benign and rarely require intervention. Pancreatic cysts in VHL do not show enhancement on imaging and do not have malignant potential, regardless of size. Diffuse cystic disease of the pancreas rarely affects endocrine function. Infrequently, cystic replacement of the normal pancreas can lead to a loss of exocrine function. When bloating, cramping, diarrhea, or abdominal pain occurs with fatty meals, enzymatic studies on the stool could be employed to determine if exocrine supplementation is indicated. Solid or mixed pancreatic lesions require specialized evaluation and treatment because they may be cystadenomas or pancreatic NETs. Most pancreatic NETs are nonfunctional, but laboratory evaluation with biochemical markers, such as chromogranin A, could be considered during the workup or the follow-up. Imaging evaluation with a contrast-enhanced computed tomography (CT) or magnetic resonance imaging (MRI) are both excellent modalities to characterize pancreatic lesions. Gallium Ga 68-DOTATATE positron emission tomography (PET)/CT has also been used to detect VHL-associated tumors. The performance metrics may depend on the lesion size and optimization of contrast administration timing. When cross-sectional imaging is inconclusive, functional imaging with nuclear medicine modalities may be useful to help diagnose metastatic disease or distinguish solid microcystic adenomas from solid pancreatic NETs. Endoscopic ultrasound is a highly sensitive modality. This procedure may be offered when intravenous contrast cannot be given or when there is concern that a lesion may be a solid microcystic serous adenoma, rather than a cancer. Tissue sampling can be performed during an endoscopic procedure, but it is rarely indicated.
Surveillance of pancreatic manifestations
Serous cystadenomas do not have malignant potential and can be safely observed. Local obstruction of the bile duct or the pancreatic duct occur rarely with these lesions. Solid pancreatic NETs have a low metastatic potential. If they are localized, small, and asymptomatic, they can be safely observed without concerns. The duration and modality for pancreatic imaging is center-dependent, but general principles include performing imaging every 1 to 2 years with the same examination method to allow meaningful comparisons. Pancreatic lesions with slow doubling times, sizes less than 3 cm, and a lack of exon 3 pathogenic variants have the most favorable outcomes. In a review of 175 VHL patients with pancreatic NETs, patients with tumors less than 1.2 cm in diameter did not develop metastases or need surgeries. Patients with larger tumors (1.2–3.0 cm in diameter) and a missense variant in VHL, as opposed to other variant types, were more likely to develop metastases or require surgical invention. Tumor size, variant type, and exon location may eventually play a role in determining surveillance in patients with VHL.
Surgical interventions for pancreatic manifestations
Pancreatic cysts rarely need surgical intervention except when they exert a mass effect. Aspiration or decortication can be considered in these rare cases. Indications for surgery on pancreatic NETs can vary, but intervention is offered to lower the risk of dissemination. A review of the natural history of pancreatic NETs shows that these tumors may demonstrate nonlinear growth characteristics. Pancreatic NETs are typically resected if they are 3 cm or larger (or ≥2 cm if the tumor is located in the head of the pancreas). Tumor enucleation is safe and effective if the lesion is not located near the pancreatic duct. If it is not safe to enucleate a lesion, a distal pancreatectomy is performed. Tumors in the head of the pancreas that are 2 cm or larger are also evaluated for resection, since larger lesions in this location are more challenging to enucleate. If the lesion is located too close to the pancreatic duct, a Whipple procedure is offered. For rare situations involving large multifocal lesions, a total pancreatectomy could be considered. After surgery, if patients develop exocrine dysfunction, enzyme supplementation may improve gastrointestinal symptoms and nutritional status.
Positive lymph nodes should be removed if they are found during surgery. Surgery is still considered for individuals with locally advanced or metastatic pancreatic NETs if significant debulking can be offered. Additionally, metastatic liver lesions can often be treated with local ablative techniques or resection in select patients with VHL.
Management of Retinal Hemangioblastomas
Interventions for retinal hemangioblastomas
Treatment of retinal hemangioblastomas includes laser treatment, photodynamic therapy, and vitrectomy. Efforts have also been made to use either local or systemic therapy.
Laser photocoagulation
Laser photocoagulation is used extensively to treat retinal hemangioblastomas in patients with VHL. A retrospective review of 304 treated retinal hemangioblastomas in 100 eyes showed that laser photocoagulation had a control rate greater than 90% and was most effective in smaller lesions up to 1 disk diameter.
Vitrectomy and retinectomy
Twenty-one patients with severe retinal detachment achieved varying degrees of visual preservation when treated with pars plana vitrectomy with posterior hyaloid detachment, epiretinal membrane dissection, and silicone oil/gas injection with retinectomy or photocoagulation/cryotherapy to remove the retinal hemangioblastoma. Pars plana vitrectomy was shown to improve or preserve visual function in 23 patients with advanced VHL eye disease. However, postoperative progression of ocular VHL disease was possibly accelerated in cases where a retinotomy was performed.
Photodynamic therapy
Photodynamic therapy reduced macular edema in a case series of two patients with bilateral retinal hemangioblastomas. These patients had minimal benefit, with little to no increase in visual acuity. In a second series of five patients, including four with VHL disease, photodynamic therapy was performed on six eyes. This therapy resulted in tumor regression/stabilization and a decrease in the amount of subretinal fluid and lipid exudates in all patients. However, stabilization or improvement of visual acuity was observed in only 50% of cases.
Intravitreal treatment
Intravitreal treatment with bevacizumab resulted in stabilization of retinal capillary hemangioblastomas for over 2 years in one case report and vision improvement in one of five eyes in a second case report. Intravitreal ranibizumab did not provide consistent benefit in a case series of five patients. Systemic bevacizumab provided marginal benefit in individual case reports. Treatment with sunitinib resulted in possible visual stabilization in three patients but with significant concomitant toxicity.
Proton therapy
In a case study in which proton therapy was used on eight eyes (in eight patients), macular edema was resolved in seven of eight patients. All treated eyes had vision preserved after a median follow-up period of 84 months.
Management of Central Nervous System (CNS) Hemangioblastomas
Surveillance of CNS hemangioblastomas
Many small lesions are found incidentally with screening, and patients may remain asymptomatic for a long time. In a study with a short-term follow-up period, 35.5% to 51% of CNS hemangioblastomas remained stable.
In a National Institutes of Health series, researchers noted that patterns of growth for CNS hemangioblastomas can vary, with saltatory growth patterns occurring most often (72%). Other growth patterns, such as linear growth (6%) and exponential growth (22%), were also observed. Determinants of growth have been assessed. Lesion location may matter, since cerebellar and brain stem lesions grow faster than those in the spinal cord or cauda equina. Approximately 12% of patients with hemangioblastomas developed peritumoral or intratumoral cysts, and 6.4% of these individuals were symptomatic and required treatment. Increased tumor burden or total tumor number detected was associated with male sex, longer follow-up time, and genotype (all P< .01). Partial germline deletions were associated with more tumors per patient than missense variants (P< .01). Other interesting findings were observed, including an increased number of tumors per year in young patients and an increased tumor growth rate in men (P< .01).
Another small series (n = 52) aimed to evaluate growth rates of CNS hemangioblastomas under surveillance. Researchers found that symptomatic presentation was the only independent predictor of growth. Since most CNS hemangioblastomas eventually grew and became symptomatic, the rate of treatment also increased, with the rates of intervention at 1, 3, and 7 years reaching 11.5%, 50.1%, and 73%, respectively.
Surgical interventions for CNS hemangioblastomas
Surgical resection of cerebellar or spinal hemangioblastomas has been the standard treatment approach. While surgical resection of tumors is generally performed prior to the onset of neurological symptoms, this varies by center and may be influenced by factors like edema, tumor location, hydrocephalus, and the tumor's growth rate. Spinal lesions are often approached posteriorly and require a laminectomy. Because patients often require multiple operations during their lifetimes, removal of support can lead to progressive spinal instability requiring stabilization/fusion. For cerebellar lesions, the surgical approach depends on the lateral orientation of the tumor, but many can be approached through a midline suboccipital incision. Preoperative embolization can be performed to reduce bleeding, but this approach depends on the surgeon's preference.
Radiation therapy for CNS hemangioblastomas
Because patients may have multiple tumors and require several surgical procedures, external beam radiation therapy has emerged as an alternative when surgical resection is not feasible. Stereotactic radiosurgery is a commonly used approach for hemangioblastoma treatment. Retrospective series have demonstrated that radiosurgery was associated with a size reduction in more than 50% of treated lesions, with a low complication rate. A prospective study at the NCI evaluated local control of treated lesions. Long-term series may be necessary to assess the effectiveness of radiosurgery, since tumors can have a saltatory growth pattern. In this series, 33% of treated subcentimeter, asymptomatic tumors progressed during follow-up. Because of concerns about long-term local control, the authors concluded that stereotactic radiosurgery should be reserved for the treatment of tumors not amenable to surgical resection. Other series have shown better outcomes. However, it is unclear if this depends on the definition of local control, which varies across studies.
Management of Endolymphatic Sac Tumors (ELSTs)
There are limited data on the management of ELSTs, consisting largely of case series detailing surgical management of sporadic and VHL-associated tumors. Because audiovestibular compromise is not dependent on tumor size and can occur with small tumors, early intervention is generally preferred. Early intervention may also minimize the risk of invasion into surrounding structures and increase the probability of complete resection. A meta-analysis assessed outcomes from 82 studies that treated 252 tumors. A limited number of patients were observed, and nearly all of them showed clinical progression, which argues against this approach. Surgery has been the main treatment for ELSTs, with complete resection the optimal choice to avoid recurrence. Preoperative embolization has been performed to reduce the risk of perioperative morbidity and hemorrhage. The surgical approach often depends on a surgeon's preference, tumor location, and the patient's baseline hearing status. Complete resection of all disease is not always feasible due to tumor extension and invasion into surrounding structures. Thus, recurrence rates for ELSTs after surgery are high (16%). While radiation is not usually a primary treatment option, it is often performed as adjuvant treatment. Data are limited on systemic therapy approaches for ELSTs.
VHL-Specific Systemic Therapy for Localized Disease
Patients with VHL often require multiple local treatments for their disease manifestations. Recurrent surgical intervention contributes to morbidity and can often cause irreversible damage to affected organs. Permanent loss of function can occur in the following organs:
- Visual impairment (retina).
- Chronic kidney disease (kidney).
- Adrenal insufficiency (adrenal glands).
- Diabetes and pancreatic exocrine deficiency (pancreas).
- Neurological complications such as motor or sensory deficits (brain and spine).
Researchers have sought a systemic therapy that can reduce or eliminate the need for local interventions. Understanding the biology of VHL has led to the development of targeted therapies that interfere with the downstream signaling cascade associated with tumorigenesis. Effective agents that target the VHL–hypoxia-inducible factor (HIF) pathway were developed to treat patients with advanced, sporadic ccRCCs. Afterward, the clinical utility of these agents was investigated in individuals with VHL who had various VHL-related manifestations.
Initial research on tanespimycin (17-AAG) therapy highlighted that some patients may not prefer intravenous administration of medication. The modest toxicity and poor tolerability associated with 17-AAG may deter healthy patients (with other surgical options) from using this treatment. Subsequently, a variety of agents targeting the vascular endothelial growth factor (VEGF) pathway have been evaluated. Most of these agents demonstrated activity against renal tumors but only modest or absent activity against CNS hemangioblastomas, pancreatic neuroendocrine tumors, and other VHL-related manifestations. A first-generation tyrosine kinase inhibitor, sunitinib, was given to patients with VHL for up to four cycles (4 weeks on, then 2 weeks off). This drug was first explored in a cohort of 15 participants with various VHL-associated manifestations, all of whom did not need immediate medical intervention. While tumors did not enlarge in these individuals (>90% of participants), toxicity (including fatigue) led to frequent sunitinib dose reductions in 10 of 15 participants. The overall response rate (ORR) was 33% (6 of 18 tumors) in localized renal tumors, demonstrating the potential for sunitinib therapy to influence the need for local intervention. Unfortunately, responses (as defined by Response Evaluation Criteria In Solid Tumors [RECIST]) were not observed in CNS hemangioblastomas. In a retrospective review of 14 patients who had received sunitinib, responses were not seen in 11 patients with cerebellar hemangioblastomas and 8 patients with spinal hemangioblastomas. Vandetanib, a tyrosine kinase inhibitor with a broad target specificity (including vascular endothelial growth factor receptor [VEGFR] and epidermal growth factor receptor [EGFR]), was evaluated in a phase II study of 37 patients. In this study, vandetanib was associated with limited activity and toxicity, which required frequent treatment interruptions and dose reductions. Pazopanib, a second-generation tyrosine kinase inhibitor with a better toxicity profile than sunitinib, was similarly studied in patients with VHL. A cohort of 31 patients was treated with pazopanib for 24 weeks with an option to continue this therapy. Responses (as defined by RECIST) were observed in 52% of renal tumors and 53% of pancreatic lesions. Both malignant pancreatic NETs and benign pancreatic serous cystadenomas were included in this study. However, there was little activity against CNS hemangioblastomas with an ORR of 4%. Pazopanib was also poorly tolerated, with only 23% of participants choosing to continue treatment beyond the initial 24-week period.
While anti-VEGF therapy is administered systemically for most VHL-associated neoplasms, in VHL-associated retinal tumors, it can be delivered directly into the eye. Intravitreally-administered pegaptanib (an anti-VEGF therapy) was evaluated in five patients with VHL-associated retinal hemangioblastomas. Only two patients were able to complete the pegaptanib therapy, and responses were not seen in the patients' primary tumors. Two patients had decreased retinal thickening and a reduced number of hard exudates. Although the U.S. Food and Drug Administration has approved pegaptanib to treat macular degeneration, it is not approved for the treatment of VHL-associated retinal lesions.
Research targeting downstream consequences of HIF upregulation (due to inactivation of the VHL gene) have had only modest success. Hence, recent efforts have focused on targeting direct consequences of VHL loss. Multiple studies have demonstrated that multiple HIF molecules differentially regulate tumorigenesis, with HIF2 being the most critical mediator. This transcription factor became a promising target for treatment, but this only became possible when a new class of agents was developed that can selectively inhibit HIF2-alpha. These agents were designed to fit in the binding pocket of HIF2-alpha's Per-ARNT-SIM (PAS)-B domain. This prevented HIF1-beta/ARNT dimerization and showed activity in laboratory models. This finding ultimately led to the development of belzutifan, an oral agent that was studied in an international phase II study of 61 participants with VHL-associated RCC across 11 centers. Unlike agents targeting the VEGF pathway, belzutifan was well-tolerated in participants and had a low rate of high-grade toxicity. The most common grade 3 adverse event was anemia (8% of patients), and most patients remained on belzutifan after this paper was published (median follow-up period, 21.8 mo). Responses were observed in 49% of renal tumors, 77% of pancreatic tumors, and 30% of CNS tumors. All retinal tumors (16/16) improved after belzutifan treatment. The FDA approved belzutifan for VHL-associated renal, pancreatic, and CNS hemangioblastomas that do not require immediate surgical intervention, based on activity and tolerability measurements from the trial. Additional research is expected to reveal the long-term efficacy and toxicity of this agent. Belzutifan is now used as an adjunct to surgical management in patients with VHL-associated tumors that are confined to an organ. It is unclear how belzutifan will be used in clinical practice and which providers will prescribe it to patients.
VHL Management During Pregnancy
Two studies have examined the effect of pregnancy on hemangioblastoma progression in patients with VHL. One study retrospectively examined the records of 29 patients with VHL from the Netherlands who became pregnant 48 times (49 newborns) between 1966 and 2010 (40% became pregnant before 1990). Imaging records were available for 31% of the pregnancies. Researchers reported that 17% of all pregnancies had VHL-related complications, including three patients with craniospinal hemangioblastomas in whom the progression score of the tumors changed significantly (P = .049) before and after pregnancy. However, findings from this study contrast those reported from a small, prospective investigation. Until a large-scale, international, prospective study is conducted, these investigations suggest using a conservative approach that includes medical surveillance during pregnancy.
References
- Goldfarb DA, Neumann HP, Penn I, et al.: Results of renal transplantation in patients with renal cell carcinoma and von Hippel-Lindau disease. Transplantation 64 (12): 1726-9, 1997.
- Fetner CD, Barilla DE, Scott T, et al.: Bilateral renal cell carcinoma in von Hippel-Lindau syndrome: treatment with staged bilateral nephrectomy and hemodialysis. J Urol 117 (4): 534-6, 1977.
- Pearson JC, Weiss J, Tanagho EA: A plea for conservation of kidney in renal adenocarcinoma associated with von Hippel-Lindau disease. J Urol 124 (6): 910-2, 1980.
- Loughlin KR, Gittes RF: Urological management of patients with von Hippel-Lindau's disease. J Urol 136 (4): 789-91, 1986.
- Steinbach F, Novick AC, Zincke H, et al.: Treatment of renal cell carcinoma in von Hippel-Lindau disease: a multicenter study. J Urol 153 (6): 1812-6, 1995.
- Walther MM, Thompson N, Linehan W: Enucleation procedures in patients with multiple hereditary renal tumors. World J Urol 13 (4): 248-50, 1995.
- Walther MM, Choyke PL, Glenn G, et al.: Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol 161 (5): 1475-9, 1999.
- Duffey BG, Choyke PL, Glenn G, et al.: The relationship between renal tumor size and metastases in patients with von Hippel-Lindau disease. J Urol 172 (1): 63-5, 2004.
- Peng X, Chen J, Wang J, et al.: Natural history of renal tumours in von Hippel-Lindau disease: a large retrospective study of Chinese patients. J Med Genet 56 (6): 380-387, 2019.
- Zhang J, Pan JH, Dong BJ, et al.: Active surveillance of renal masses in von Hippel-Lindau disease: growth rates and clinical outcome over a median follow-up period of 56 months. Fam Cancer 11 (2): 209-14, 2012.
- Jilg CA, Neumann HP, Gläsker S, et al.: Growth kinetics in von Hippel-Lindau-associated renal cell carcinoma. Urol Int 88 (1): 71-8, 2012.
- Neumann HP, Bender BU, Berger DP, et al.: Prevalence, morphology and biology of renal cell carcinoma in von Hippel-Lindau disease compared to sporadic renal cell carcinoma. J Urol 160 (4): 1248-54, 1998.
- Fadahunsi AT, Sanford T, Linehan WM, et al.: Feasibility and outcomes of partial nephrectomy for resection of at least 20 tumors in a single renal unit. J Urol 185 (1): 49-53, 2011.
- Choyke PL, Pavlovich CP, Daryanani KD, et al.: Intraoperative ultrasound during renal parenchymal sparing surgery for hereditary renal cancers: a 10-year experience. J Urol 165 (2): 397-400, 2001.
- Bratslavsky G, Liu JJ, Johnson AD, et al.: Salvage partial nephrectomy for hereditary renal cancer: feasibility and outcomes. J Urol 179 (1): 67-70, 2008.
- Johnson A, Sudarshan S, Liu J, et al.: Feasibility and outcomes of repeat partial nephrectomy. J Urol 180 (1): 89-93; discussion 93, 2008.
- Shuch B, Linehan WM, Bratslavsky G: Repeat partial nephrectomy: surgical, functional and oncological outcomes. Curr Opin Urol 21 (5): 368-75, 2011.
- Gill IS, Novick AC, Soble JJ, et al.: Laparoscopic renal cryoablation: initial clinical series. Urology 52 (4): 543-51, 1998.
- McGovern FJ, Wood BJ, Goldberg SN, et al.: Radio frequency ablation of renal cell carcinoma via image guided needle electrodes. J Urol 161 (2): 599-600, 1999.
- Campbell SC, Novick AC, Belldegrun A, et al.: Guideline for management of the clinical T1 renal mass. J Urol 182 (4): 1271-9, 2009.
- Walther MC, Shawker TH, Libutti SK, et al.: A phase 2 study of radio frequency interstitial tissue ablation of localized renal tumors. J Urol 163 (5): 1424-7, 2000.
- Shingleton WB, Sewell PE: Percutaneous renal cryoablation of renal tumors in patients with von Hippel-Lindau disease. J Urol 167 (3): 1268-70, 2002.
- Park BK, Kim CK: Percutaneous radio frequency ablation of renal tumors in patients with von Hippel-Lindau disease: preliminary results. J Urol 183 (5): 1703-7, 2010.
- Chan VW, Lenton J, Smith J, et al.: Multimodal image-guided ablation on management of renal cancer in Von-Hippel-Lindau syndrome patients from 2004 to 2021 at a specialist centre: A longitudinal observational study. Eur J Surg Oncol 48 (3): 672-679, 2022.
- Joly D, Méjean A, Corréas JM, et al.: Progress in nephron sparing therapy for renal cell carcinoma and von Hippel-Lindau disease. J Urol 185 (6): 2056-60, 2011.
- Yang B, Autorino R, Remer EM, et al.: Probe ablation as salvage therapy for renal tumors in von Hippel-Lindau patients: the Cleveland Clinic experience with 3 years follow-up. Urol Oncol 31 (5): 686-92, 2013.
- Nguyen CT, Lane BR, Kaouk JH, et al.: Surgical salvage of renal cell carcinoma recurrence after thermal ablative therapy. J Urol 180 (1): 104-9; discussion 109, 2008.
- Karam JA, Wood CG, Compton ZR, et al.: Salvage surgery after energy ablation for renal masses. BJU Int 115 (1): 74-80, 2015.
- Kowalczyk KJ, Hooper HB, Linehan WM, et al.: Partial nephrectomy after previous radio frequency ablation: the National Cancer Institute experience. J Urol 182 (5): 2158-63, 2009.
- Shuch B, Singer EA, Bratslavsky G: The surgical approach to multifocal renal cancers: hereditary syndromes, ipsilateral multifocality, and bilateral tumors. Urol Clin North Am 39 (2): 133-48, v, 2012.
- Dominguez-Escrig JL, Sahadevan K, Johnson P: Cryoablation for small renal masses. Adv Urol : 479495, 2008.
- Aron M, Gill IS: Minimally invasive nephron-sparing surgery (MINSS) for renal tumours. Part II: probe ablative therapy. Eur Urol 51 (2): 348-57, 2007.
- Sanford T, Gomella PT, Siddiqui R, et al.: Long term outcomes for patients with von Hippel-Lindau and Pheochromocytoma: defining the role of active surveillance. Urol Oncol 39 (2): 134.e1-134.e8, 2021.
- Benhammou JN, Boris RS, Pacak K, et al.: Functional and oncologic outcomes of partial adrenalectomy for pheochromocytoma in patients with von Hippel-Lindau syndrome after at least 5 years of followup. J Urol 184 (5): 1855-9, 2010.
- Baghai M, Thompson GB, Young WF, et al.: Pheochromocytomas and paragangliomas in von Hippel-Lindau disease: a role for laparoscopic and cortical-sparing surgery. Arch Surg 137 (6): 682-8; discussion 688-9, 2002.
- Brauckhoff M, Gimm O, Thanh PN, et al.: Critical size of residual adrenal tissue and recovery from impaired early postoperative adrenocortical function after subtotal bilateral adrenalectomy. Surgery 134 (6): 1020-7; discussion 1027-8, 2003.
- Walther MM, Keiser HR, Choyke PL, et al.: Management of hereditary pheochromocytoma in von Hippel-Lindau kindreds with partial adrenalectomy. J Urol 161 (2): 395-8, 1999.
- Fallon SC, Feig D, Lopez ME, et al.: The utility of cortical-sparing adrenalectomy in pheochromocytomas associated with genetic syndromes. J Pediatr Surg 48 (6): 1422-5, 2013.
- Roukounakis N, Dimas S, Kafetzis I, et al.: Is preservation of the adrenal vein mandatory in laparoscopic adrenal-sparing surgery? JSLS 11 (2): 215-8, 2007 Apr-Jun.
- Timmers HJ, Gimenez-Roqueplo AP, Mannelli M, et al.: Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocr Relat Cancer 16 (2): 391-400, 2009.
- Vargas HI, Kavoussi LR, Bartlett DL, et al.: Laparoscopic adrenalectomy: a new standard of care. Urology 49 (5): 673-8, 1997.
- Perrier ND, Kennamer DL, Bao R, et al.: Posterior retroperitoneoscopic adrenalectomy: preferred technique for removal of benign tumors and isolated metastases. Ann Surg 248 (4): 666-74, 2008.
- Dickson PV, Jimenez C, Chisholm GB, et al.: Posterior retroperitoneoscopic adrenalectomy: a contemporary American experience. J Am Coll Surg 212 (4): 659-65; discussion 665-7, 2011.
- Binderup ML, Bisgaard ML, Harbud V, et al.: Von Hippel-Lindau disease (vHL). National clinical guideline for diagnosis and surveillance in Denmark. 3rd edition. Dan Med J 60 (12): B4763, 2013.
- Kruizinga RC, Sluiter WJ, de Vries EG, et al.: Calculating optimal surveillance for detection of von Hippel-Lindau-related manifestations. Endocr Relat Cancer 21 (1): 63-71, 2014.
- Keutgen XM, Hammel P, Choyke PL, et al.: Evaluation and management of pancreatic lesions in patients with von Hippel-Lindau disease. Nat Rev Clin Oncol 13 (9): 537-49, 2016.
- Shell J, Tirosh A, Millo C, et al.: The utility of 68Gallium-DOTATATE PET/CT in the detection of von Hippel-Lindau disease associated tumors. Eur J Radiol 112: 130-135, 2019.
- Sadowski SM, Weisbrod AB, Ellis R, et al.: Prospective evaluation of the clinical utility of 18-fluorodeoxyglucose PET CT scanning in patients with von hippel-lindau-associated pancreatic lesions. J Am Coll Surg 218 (5): 997-1003, 2014.
- Blansfield JA, Choyke L, Morita SY, et al.: Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs). Surgery 142 (6): 814-8; discussion 818.e1-2, 2007.
- Tirosh A, Sadowski SM, Linehan WM, et al.: Association of VHL Genotype With Pancreatic Neuroendocrine Tumor Phenotype in Patients With von Hippel-Lindau Disease. JAMA Oncol 4 (1): 124-126, 2018.
- Weisbrod AB, Kitano M, Thomas F, et al.: Assessment of tumor growth in pancreatic neuroendocrine tumors in von Hippel Lindau syndrome. J Am Coll Surg 218 (2): 163-9, 2014.
- Libutti SK, Choyke PL, Bartlett DL, et al.: Pancreatic neuroendocrine tumors associated with von Hippel Lindau disease: diagnostic and management recommendations. Surgery 124 (6): 1153-9, 1998.
- Krivosic V, Kamami-Levy C, Jacob J, et al.: Laser photocoagulation for peripheral retinal capillary hemangioblastoma in von Hippel-Lindau disease. Ophthalmol Retina 1 (1): 59-67, 2017. Also available online. Last accessed January 3, 2024.
- Gaudric A, Krivosic V, Duguid G, et al.: Vitreoretinal surgery for severe retinal capillary hemangiomas in von hippel-lindau disease. Ophthalmology 118 (1): 142-9, 2011.
- Krzystolik K, Stopa M, Kuprjanowicz L, et al.: PARS PLANA VITRECTOMY IN ADVANCED CASES OF VON HIPPEL-LINDAU EYE DISEASE. Retina 36 (2): 325-34, 2016.
- Papastefanou VP, Pilli S, Stinghe A, et al.: Photodynamic therapy for retinal capillary hemangioma. Eye (Lond) 27 (3): 438-42, 2013.
- Sachdeva R, Dadgostar H, Kaiser PK, et al.: Verteporfin photodynamic therapy of six eyes with retinal capillary haemangioma. Acta Ophthalmol 88 (8): e334-40, 2010.
- Ach T, Thiemeyer D, Hoeh AE, et al.: Intravitreal bevacizumab for retinal capillary haemangioma: longterm results. Acta Ophthalmol 88 (4): e137-8, 2010.
- Slim E, Antoun J, Kourie HR, et al.: Intravitreal bevacizumab for retinal capillary hemangioblastoma: A case series and literature review. Can J Ophthalmol 49 (5): 450-7, 2014.
- Wong WT, Liang KJ, Hammel K, et al.: Intravitreal ranibizumab therapy for retinal capillary hemangioblastoma related to von Hippel-Lindau disease. Ophthalmology 115 (11): 1957-64, 2008.
- von Buelow M, Pape S, Hoerauf H: Systemic bevacizumab treatment of a juxtapapillary retinal haemangioma. Acta Ophthalmol Scand 85 (1): 114-6, 2007.
- Wackernagel W, Lackner EM, Pilz S, et al.: von Hippel-Lindau disease: treatment of retinal haemangioblastomas by targeted therapy with systemic bevacizumab. Acta Ophthalmol 88 (7): e271-2, 2010.
- Knickelbein JE, Jacobs-El N, Wong WT, et al.: Systemic Sunitinib Malate Treatment for Advanced Juxtapapillary Retinal Hemangioblastomas Associated with von Hippel-Lindau Disease. Ophthalmol Retina 1 (3): 181-187, 2017 May-Jun.
- Seibel I, Cordini D, Hager A, et al.: Long-term results after proton beam therapy for retinal papillary capillary hemangioma. Am J Ophthalmol 158 (2): 381-6, 2014.
- Byun J, Yoo HJ, Kim JH, et al.: Growth rate and fate of untreated hemangioblastomas: clinical assessment of the experience of a single institution. J Neurooncol 144 (1): 147-154, 2019.
- Lonser RR, Butman JA, Huntoon K, et al.: Prospective natural history study of central nervous system hemangioblastomas in von Hippel-Lindau disease. J Neurosurg 120 (5): 1055-62, 2014.
- Harati A, Satopää J, Mahler L, et al.: Early microsurgical treatment for spinal hemangioblastomas improves outcome in patients with von Hippel-Lindau disease. Surg Neurol Int 3: 6, 2012.
- Asthagiri AR, Mehta GU, Butman JA, et al.: Long-term stability after multilevel cervical laminectomy for spinal cord tumor resection in von Hippel-Lindau disease. J Neurosurg Spine 14 (4): 444-52, 2011.
- Das JM, Kesavapisharady K, Sadasivam S, et al.: Microsurgical Treatment of Sporadic and von Hippel-Lindau Disease Associated Spinal Hemangioblastomas: A Single-Institution Experience. Asian Spine J 11 (4): 548-555, 2017.
- Kano H, Shuto T, Iwai Y, et al.: Stereotactic radiosurgery for intracranial hemangioblastomas: a retrospective international outcome study. J Neurosurg 122 (6): 1469-78, 2015.
- Asthagiri AR, Mehta GU, Zach L, et al.: Prospective evaluation of radiosurgery for hemangioblastomas in von Hippel-Lindau disease. Neuro Oncol 12 (1): 80-6, 2010.
- Liebenow B, Tatter A, Dezarn WA, et al.: Gamma Knife Stereotactic Radiosurgery favorably changes the clinical course of hemangioblastoma growth in von Hippel-Lindau and sporadic patients. J Neurooncol 142 (3): 471-478, 2019.
- Tang JD, Grady AJ, Nickel CJ, et al.: Systematic Review of Endolymphatic Sac Tumor Treatment and Outcomes. Otolaryngol Head Neck Surg 168 (3): 282-290, 2023.
- Linehan WM, Pinto PA, Bratslavsky G, et al.: Hereditary kidney cancer: unique opportunity for disease-based therapy. Cancer 115 (10 Suppl): 2252-61, 2009.
- Shuch B: HIF2 Inhibition for von-Hippel Lindau Associated Kidney Cancer: Will Urology Lead or Follow? Urol Oncol 39 (5): 277-280, 2021.
- Jonasch E, McCutcheon IE, Waguespack SG, et al.: Pilot trial of sunitinib therapy in patients with von Hippel-Lindau disease. Ann Oncol 22 (12): 2661-6, 2011.
- Roma A, Maruzzo M, Basso U, et al.: First-Line sunitinib in patients with renal cell carcinoma (RCC) in von Hippel-Lindau (VHL) disease: clinical outcome and patterns of radiological response. Fam Cancer 14 (2): 309-16, 2015.
- Stamatakis L, Shuch B, Singer EA, et al.: Phase II trial of vandetanib in Von Hippel-Lindau-associated renal cell carcinoma. J Clin Oncol 31 (15 Suppl): 4584, 2013. Also available online. Last accessed January 3, 2024.
- Jonasch E, McCutcheon IE, Gombos DS, et al.: Pazopanib in patients with von Hippel-Lindau disease: a single-arm, single-centre, phase 2 trial. Lancet Oncol 19 (10): 1351-1359, 2018.
- Dahr SS, Cusick M, Rodriguez-Coleman H, et al.: Intravitreal anti-vascular endothelial growth factor therapy with pegaptanib for advanced von Hippel-Lindau disease of the retina. Retina 27 (2): 150-8, 2007.
- Kondo K, Kim WY, Lechpammer M, et al.: Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol 1 (3): E83, 2003.
- Courtney KD, Infante JR, Lam ET, et al.: Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2α Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J Clin Oncol 36 (9): 867-874, 2018.
- Jonasch E, Donskov F, Iliopoulos O, et al.: Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N Engl J Med 385 (22): 2036-2046, 2021.
- Frantzen C, Kruizinga RC, van Asselt SJ, et al.: Pregnancy-related hemangioblastoma progression and complications in von Hippel-Lindau disease. Neurology 79 (8): 793-6, 2012.
- Ye DY, Bakhtian KD, Asthagiri AR, et al.: Effect of pregnancy on hemangioblastoma development and progression in von Hippel-Lindau disease. J Neurosurg 117 (5): 818-24, 2012.
Prognosis
Historically, patients with von Hippel-Lindau disease (VHL) had poor survival when compared with the general population, because of the morbidity and mortality of the various disease manifestations and the resultant management. With improved understanding of the disease and modern management strategies, the outcomes may be improving. A Danish study compared the life expectancy of individuals with VHL with that of their unaffected siblings; this demonstrated a median survival of 67 years in men and 60 years in women with VHL disease. A second study from China involving 340 patients with VHL disease estimated a median life expectancy of 62 years of age.
In the past, metastatic renal cell carcinoma (RCC) has caused about one-third of deaths in patients with VHL and, in some reports, it was the leading cause of death in VHL patients. With increased surveillance of pathogenic variant–positive individuals, the RCC mortality rate is thought to have diminished significantly because of adherence to RCC treatment recommendations including the 3 cm rule. The study from China demonstrated that 97% of deaths in patients with VHL were disease related, with central nervous system (CNS) hemangioblastoma and RCC accounting for 67.7% and 27.8% of deaths, respectively. The Danish study demonstrated 79% of deaths were VHL related with 51% attributed to CNS hemangioblastoma and 36% attributed to RCC.
Morbidity and mortality in VHL vary and are influenced by the individual and the family’s VHL phenotype (e.g., type 1, 2A, 2B, or 2C). For more information, see the Genotype-phenotype correlations section. The Danish study reported an increased hazard ratio for death in women, but this was not observed in the Chinese series. The age and type of presentation may influence outcome; this is perhaps reflective of disease aggressiveness. Genotype did not have an effect on survival probability in the Danish series but, for the Chinese study, those with type 2 disease had improved survival (compared with type 1) and those with a missense variant had improved survival (compared with a truncating variant).
References
- Maher ER, Yates JR, Harries R, et al.: Clinical features and natural history of von Hippel-Lindau disease. Q J Med 77 (283): 1151-63, 1990.
- Binderup ML, Jensen AM, Budtz-Jørgensen E, et al.: Survival and causes of death in patients with von Hippel-Lindau disease. J Med Genet 54 (1): 11-18, 2017.
- Wang JY, Peng SH, Li T, et al.: Risk factors for survival in patients with von Hippel-Lindau disease. J Med Genet 55 (5): 322-328, 2018.
- Lamiell JM, Salazar FG, Hsia YE: von Hippel-Lindau disease affecting 43 members of a single kindred. Medicine (Baltimore) 68 (1): 1-29, 1989.
- Horton WA, Wong V, Eldridge R: Von Hippel-Lindau disease: clinical and pathological manifestations in nine families with 50 affected members. Arch Intern Med 136 (7): 769-77, 1976.
- Neumann HP: Basic criteria for clinical diagnosis and genetic counselling in von Hippel-Lindau syndrome. Vasa 16 (3): 220-6, 1987.
Future Directions
Currently, the renal manifestations of von Hippel-Lindau disease (VHL) are generally managed surgically or with thermal ablation. There is a clear need for better management strategies and development of targeted systemic therapy. These will include defining the molecular biology and genetics of kidney cancer formation, which may lead to the development of effective prevention or early intervention therapies. In addition, the evolving understanding of the molecular biology of established kidney cancers may provide opportunities to phenotypically normalize the cancer by modulating residual VHLgene function, identifying new targets for treatment, and discovering synthetic lethal strategies that can effectively eradicate renal cell carcinoma.
Latest Updates to This Summary (04/05/2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
This summary was extensively revised.
This summary is written and maintained by the PDQ Cancer Genetics Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the genetics of von Hippel-Lindau disease. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Cancer Genetics Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Von Hippel-Lindau Disease are:
- Alexandra Perez Lebensohn, MS, CGC (National Cancer Institute)
- Brian Matthew Shuch, MD (UCLA Health)
- Ramaprasad Srinivasan, MD, PhD (National Cancer Institute)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Cancer Genetics Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Cancer Genetics Editorial Board. PDQ Von Hippel-Lindau Disease. Bethesda, MD: National Cancer Institute. Updated
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
The information in these summaries should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.