National Cancer Institute
Post Date: Nov 5, 2023
Genetics of Skin Cancer includes information about genes and hereditary syndromes associated with basal cell, squamous cell, and melanoma skin cancer. Get comprehensive information about the genetics of skin cancer and interventions in this summary for clinicians.
Genetics of Skin Cancer
Executive Summary
This executive summary reviews the topics covered in this PDQ summary on the genetics of skin cancer, with hyperlinks to detailed sections below that describe the evidence on each topic.
- Inheritance and Risk
More than 100 types of tumors are clinically apparent on the skin. Many are known to have familial and/or inherited components, either in isolation or as part of a syndrome with other features. Basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) are two of the most common malignancies in the United States and are often caused by sun exposure, although several hereditary syndromes and genes are also associated with an increased risk of developing these cancers. Melanoma (which is sometimes referred to as cutaneous melanoma) is a less common type of skin cancer, but 5% to 10% of all melanomas arise in multiple-case families and can be inherited in an autosomal dominant fashion. Melanoma is the most lethal of the common skin cancers.
- Associated Genes and Syndromes
Several genes and hereditary syndromes are associated with the development of skin cancer:
- Basal cell carcinoma - Basal cell nevus syndrome (BCNS, caused by pathogenic variants in PTCH1 and PTCH2) is associated with increased BCC risk.
- Squamous cell carcinoma - Syndromes such as oculocutaneous albinism, epidermolysis bullosa, and Fanconi anemia are associated with increased SCC risk.
- Melanoma - CDKN2A is a major germlinetumor suppressor gene that is associated with increased melanoma risk. Pathogenic variants in CDKN2A may account for 35% to 40% of all familial melanomas. Germline pathogenic variants in several other genes (i.e., CDK4, MITF, and BAP1) are also associated with increased melanoma risk.
An autosomal recessive disease, called xeroderma pigmentosum (XP), is associated with increased BCC, SCC, and melanoma risks.
Genome-wide association studies show promise for identifying common, low-penetrance susceptibility alleles for many complex diseases, including melanoma, but the clinical utility of these findings remains uncertain.
- Clinical Management
Risk-reducing strategies for individuals with an increased hereditary predispositions to skin cancer are similar to recommendations for those in the general population. These recommendations include sun avoidance, use of sunscreen, use of sun-protective clothing, and avoidance of tanning beds. Chemopreventive agents such as isotretinoin and acitretin have been studied for the treatment of BCCs in patients with BCNS and XP and are associated with a significant decrease in the number of tumors per year. Vismodegib has also shown promise in reducing the per-patient annual rate of new BCCs requiring surgery among patients with BCNS. Isotretinoin has also been shown to reduce SCC incidence among patients with XP.
Treatment of hereditary skin cancers is similar to the treatment of sporadic skin cancers. One study in an XP population found therapeutic use of fluorouracil (5-FU) to be efficacious, particularly in the treatment of extensive lesions. In addition to its role as a therapeutic and potential chemopreventive agent, vismodegib is also being studied for potential palliative effects for keratocystic odontogenic tumors in patients with BCNS.
- Psychosocial and Behavioral Issues
Most of the psychosocial literature about hereditary skin cancers has focused on patients with familial melanoma. In individuals at risk of familial melanoma, psychosocial factors influence decisions about genetic testing for inherited cancer risk and risk-management strategies. Interest in genetic testing for pathogenic variants in CDKN2A is generally high. Perceived benefits among individuals with a strong family history of melanoma include information about the risk of melanoma for themselves and their children and increased motivation for sun-protective behavior. A number of studies have examined risk-reducing and early-detection behaviors in individuals with a family history of melanoma. Overall, these studies indicate inconsistent adoption and maintenance of these behaviors. Intervention studies have targeted knowledge about melanoma, sun protection, and screening behaviors in family members of patients with melanoma, with mixed results. Research is ongoing to better understand and address psychosocial and behavioral issues in high-risk families.
Introduction
Many of the medical and scientific terms used in this summary are found in the NCI Dictionary of Genetics Terms. When a linked term is clicked, the definition will appear in a separate window.
A concerted effort is being made within the genetics community to shift terminology used to describe genetic variation. The shift is to use the term “variant” rather than the term “mutation” to describe a difference that exists between the person or group being studied and the reference sequence, particularly for differences that exist in the germline. Variants can then be further classified as benign (harmless), likely benign, of uncertain significance, likely pathogenic, or pathogenic (disease causing). Throughout this summary, we will use the term pathogenic variant to describe a disease-causing mutation. For more information about variant classification, see Cancer Genetics Overview.
Many of the genes and conditions described in this summary are found in the Online Mendelian Inheritance in Man (OMIM) catalog. For more information, see OMIM.
Structure of the Skin
The genetics of skin cancer is an extremely broad topic. More than 100 types of tumors are clinically apparent on the skin; many of them have familial components, either in isolation or as part of a syndrome with other features. This is, in part, because the skin itself is a complex organ made up of multiple cell types. Furthermore, many of these cell types can undergo malignant transformation at various points in their differentiation, leading to tumors with distinct histology and dramatically different biological behaviors, such as squamous cell carcinoma (SCC) and basal cell cancer (BCC). These have been called nonmelanoma skin cancers or keratinocyte cancers.
Figure 1 is a simple diagram of normal skin structure. It also indicates the major cell types that are normally found in each compartment. Broadly speaking, there are two large compartments—the avascular epidermis and the vascular dermis—with many cell types distributed in a connective tissue matrix, largely created by fibroblasts.
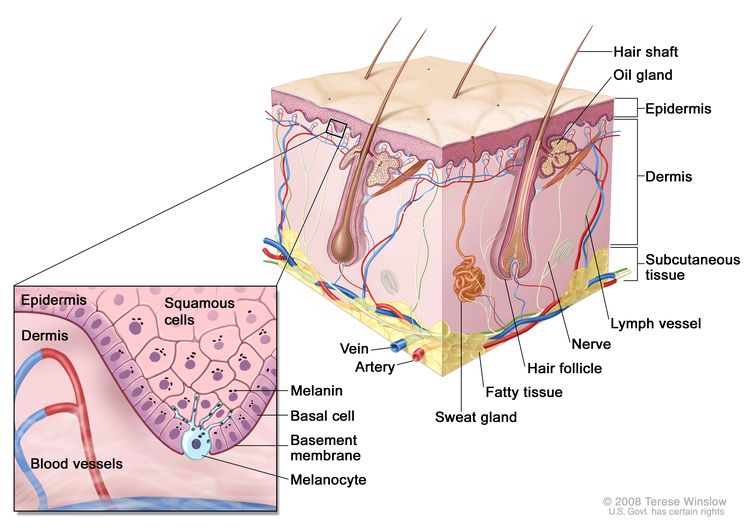 Figure 1. Schematic representation of normal skin. The relatively avascular epidermis houses basal cell keratinocytes and squamous epithelial keratinocytes, the source cells for BCC and SCC, respectively. Melanocytes are also present in normal skin and serve as the source cell for melanoma. The separation between epidermis and dermis occurs at the basement membrane zone, located just inferior to the basal cell keratinocytes.
Figure 1. Schematic representation of normal skin. The relatively avascular epidermis houses basal cell keratinocytes and squamous epithelial keratinocytes, the source cells for BCC and SCC, respectively. Melanocytes are also present in normal skin and serve as the source cell for melanoma. The separation between epidermis and dermis occurs at the basement membrane zone, located just inferior to the basal cell keratinocytes.
The outer layer or epidermis is made primarily of keratinocytes but has several other minor cell populations. The bottom layer is formed of basal keratinocytes abutting the basement membrane, along with interspersed melanocytes. The basement membrane is formed from products of keratinocytes and dermal fibroblasts, such as collagen and laminin, and is an important anatomical and functional structure. Basal keratinocytes lose contact with the basement membrane as they divide. As basal keratinocytes migrate toward the skin's surface, they progressively differentiate, lose their nuclei, and form the spinous cell layer; the granular cell layer; and the keratinized outer layer, or stratum corneum, which serves as a protective covering of the body.
The true cytologic origin of BCC is unclear. BCC and basal cell keratinocytes share many histological similarities, as is reflected in the name. Alternatively, the outer root sheath cells of the hair follicle have also been proposed as the cell of origin for BCC. This is suggested by the fact that BCCs occur predominantly on hair-bearing skin. BCCs rarely metastasize but can invade tissue locally or regionally, sometimes traveling along nerves.
Some debate remains about the origin of SCC; however, these cancers are likely derived from epidermal stem cells associated with the hair follicle. A variety of tissues, such as the lung and the uterine cervix, can give rise to SCCs. This cancer has somewhat differing behavior depending on its tissue source. Even in cancer derived from the skin, SCC from different anatomic locations can have differing levels of aggressiveness; for example, SCC from glabrous (smooth, hairless) sun-exposed skin has a lower metastatic rate than SCC arising from the vermillion border of the lip or from scars.
Additionally, in the epidermal compartment, melanocytes distribute singly along the basement membrane and can undergo malignant transformation into melanoma. Melanocytes are derived from neural crest cells and migrate to the epidermal compartment near the eighth week of gestational age. Melanocytes contain melanin, which is packaged into melanosomes and transported to nearby keratinocytes to induce pigmentation of the skin. Melanin provides a barrier for the nuclei of keratinocytes against ultraviolet radiation and also plays a role in the immune system.
Langerhans cells, or dendritic cells, are another cell type in the epidermis and have a primary function of antigen presentation. These cells reside in the skin for an extended period of time and respond to different stimuli, such as ultraviolet radiation or topical steroids, which cause them to migrate out of the skin.
The dermis is largely composed of an extracellular matrix. Prominent cell types and organelles in this compartment are fibroblasts, endothelial cells, smooth muscle cells, transient immune system cells, blood vessels, and nerves. When malignant transformation occurs, fibroblasts form fibrosarcomas and endothelial cells form angiosarcomas, Kaposi sarcoma, or other vascular tumors. There are a number of immune cell types that move in and out of the skin to blood vessels and lymphatics; these include mast cells, lymphocytes, mononuclear cells, histiocytes, and granulocytes. These cells can increase in number in inflammatory diseases and can form tumors within the skin. For example, urticaria pigmentosa is a condition that arises from mast cells and is occasionally associated with mast cell leukemia; cutaneous T-cell lymphoma is often confined to the skin throughout its course. Overall, 10% of leukemias and lymphomas have prominent expression in the skin.
Epidermal appendages are also found in the dermal compartment. These are derivatives of the epidermal keratinocytes, such as hair follicles, sweat glands, and the sebaceous glands associated with the hair follicles. These structures are generally formed in the first and second trimesters of fetal development. These can form a large variety of benign or malignant tumors with diverse biological behaviors. Several of these tumors are associated with familial syndromes. Overall, there are dozens of different histological subtypes of these tumors associated with individual components of the adnexal structures.
Finally, the subcutis is a layer that extends below the dermis with varying depth, depending on the anatomic location. This deeper boundary can include muscle, fascia, bone, or cartilage. The subcutis can be affected by inflammatory conditions such as panniculitis and malignancies such as liposarcoma.
These compartments give rise to their own malignancies but are also the region of immediate adjacent spread of localized skin cancers from other compartments. The boundaries of each skin compartment are used to define the staging of skin cancers. For example, an in situ melanoma is confined to the epidermis. Once the cancer crosses the basement membrane into the dermis, it is invasive. Internal malignancies also commonly metastasize to the skin. The dermis and subcutis are the most common locations, but the epidermis can also be involved in conditions such as Pagetoid breast cancer.
Function of the Skin
The skin has a wide variety of functions. First, the skin is an important barrier preventing extensive water and temperature loss and providing protection against minor abrasions. These functions can be aberrantly regulated in cancer. For example, in the erythroderma (extensive reddening of the skin) associated with severe sunburn, alterations in the regulations of body temperature can result in profound heat loss.
Second, the skin has important adaptive and innate immunity functions. In adaptive immunity, antigen-presenting cells engender T-cell responses consisting of increased levels of helper T cells (TH)1, TH2, or TH17. In innate immunity, the immune system produces numerous peptides with antibacterial and antifungal capacity. Even small breaks in the skin can potentially lead to infection. The skin-associated lymphoid tissue is one of the largest arms of the immune system and has a role in the prevention of infection. It may also be important in immune surveillance against cancer. Immunosuppression, such as when it is induced intentionally after solid-organ transplantation to reduce the risk of transplanted organ rejection, is a significant risk factor for skin cancer. The skin is significant for communication through facial expression and hand movements. Unfortunately, areas of specialized function, such as the area around the eyes and ears, are common places for cancer to occur. Even small cancers in these areas can lead to reconstructive challenges and have significant cosmetic and social ramifications.
Clinical Presentation of Skin Cancers
While the appearance of any one skin cancer can vary, there are general physical presentations that can be used in screening. BCCs most commonly have a pearly rim or can appear somewhat eczematous (for more information, see Figure 2 and Figure 3). They often ulcerate (for more information, see Figure 2). SCCs frequently have a thick keratin top layer (for more information, see Figure 4). Both BCCs and SCCs are associated with a history of sun-damaged skin. Melanomas are characterized by dark pigment with asymmetry, border irregularity, color variation, a diameter of more than 6 mm, and evolution (ABCDE criteria). For more information about ABCDE criteria, see What Does Melanoma Look Like? on NCI's website. Photographs representing typical clinical presentations of these cancers are shown below.
Basal cell carcinomas
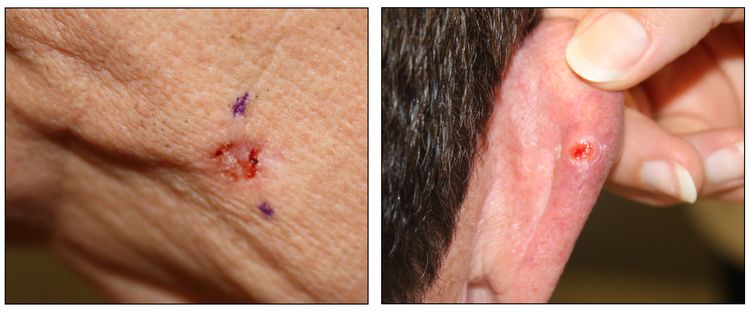 Figure 2. Ulcerated basal cell carcinoma (left panel) and ulcerated basal cell carcinoma with characteristic pearly rim (right panel).
Figure 2. Ulcerated basal cell carcinoma (left panel) and ulcerated basal cell carcinoma with characteristic pearly rim (right panel).
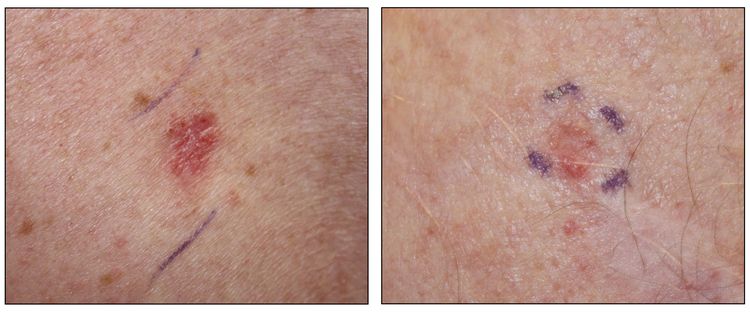 Figure 3. Superficial basal cell carcinoma (left panel) and nodular basal cell carcinoma (right panel).
Figure 3. Superficial basal cell carcinoma (left panel) and nodular basal cell carcinoma (right panel).
Squamous cell carcinomas
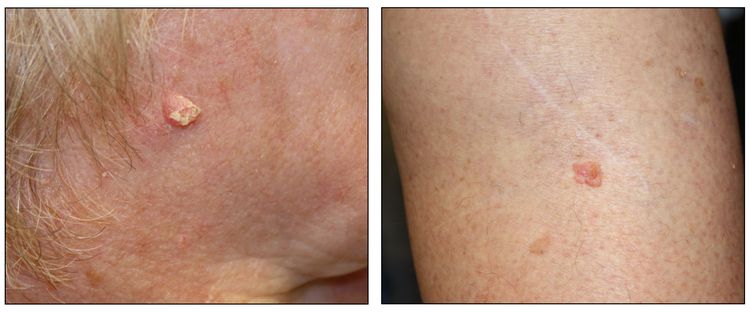 Figure 4. Squamous cell carcinoma on the face with thick keratin top layer (left panel) and squamous cell carcinoma on the leg (right panel).
Figure 4. Squamous cell carcinoma on the face with thick keratin top layer (left panel) and squamous cell carcinoma on the leg (right panel).
Melanomas
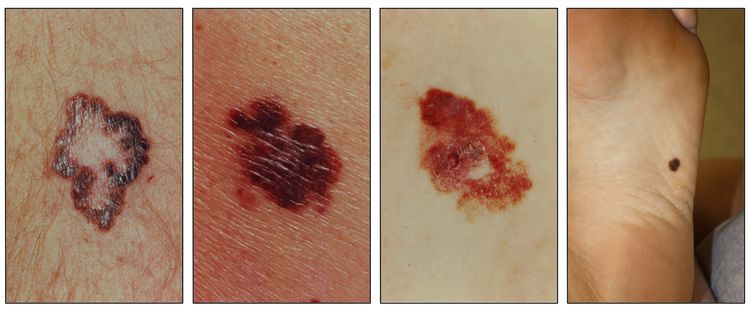 Figure 5. Melanomas with characteristic asymmetry, border irregularity, color variation, and large diameter.
Figure 5. Melanomas with characteristic asymmetry, border irregularity, color variation, and large diameter.
References
- Vandergriff TW, Bergstresser PR: Anatomy and physiology. In: Bolognia JL, Jorizzo JL, Schaffer JV: Dermatology. 3rd ed. Elsevier Saunders, 2012, pp 43-54.
- Schirren CG, Rütten A, Kaudewitz P, et al.: Trichoblastoma and basal cell carcinoma are neoplasms with follicular differentiation sharing the same profile of cytokeratin intermediate filaments. Am J Dermatopathol 19 (4): 341-50, 1997.
- Soyer HP, Rigel DS, Wurm EM: Actinic keratosis, basal cell carcinoma and squamous cell carcinoma. In: Bolognia JL, Jorizzo JL, Schaffer JV: Dermatology. 3rd ed. Elsevier Saunders, 2012, pp 1773-93.
- Lapouge G, Youssef KK, Vokaer B, et al.: Identifying the cellular origin of squamous skin tumors. Proc Natl Acad Sci U S A 108 (18): 7431-6, 2011.
- Lin JY, Fisher DE: Melanocyte biology and skin pigmentation. Nature 445 (7130): 843-50, 2007.
- Koster MI, Loomis CA, Koss TK, et al.: Skin development and maintenance. In: Bolognia JL, Jorizzo JL, Schaffer JV: Dermatology. 3rd ed. Elsevier Saunders, 2012, pp 55-64.
- Kamino H, Reddy VB, Pui J: Fibrous and fibrohistiocytic proliferations of the skin and tendons. In: Bolognia JL, Jorizzo JL, Schaffer JV: Dermatology. 3rd ed. Elsevier Saunders, 2012, pp 1961-77.
- McCalmont TH: Adnexal neoplasms. In: Bolognia JL, Jorizzo JL, Schaffer JV: Dermatology. 3rd ed. Elsevier Saunders, 2012, pp 1829-50.
- Kaddu S, Kohler S: Muscle, adipose and cartilage neoplasms. In: Bolognia JL, Jorizzo JL, Schaffer JV: Dermatology. 3rd ed. Elsevier Saunders, 2012, pp 1979-92.
- Harrington LE, Mangan PR, Weaver CT: Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Curr Opin Immunol 18 (3): 349-56, 2006.
Basal Cell Carcinoma
Introduction
Basal cell carcinoma (BCC) is the most common malignancy in people of European descent, with an associated lifetime risk of 30%. While exposure to ultraviolet (UV) radiation is the risk factor most closely linked to the development of BCC, other environmental factors (such as ionizing radiation, chronic arsenic ingestion, and immunosuppression) and genetic factors (such as family history, skin type, and genetic syndromes) also potentially contribute to carcinogenesis. In contrast to melanoma, metastatic spread of BCC is very rare and typically arises from large tumors that have evaded medical treatment for extended periods of time. BCCs can invade tissue locally or regionally, sometimes following along nerves. With early detection, the prognosis for BCC is excellent.
Risk Factors for Basal Cell Carcinoma
This section focuses on risk factors in individuals at increased hereditary risk of developing BCC. For more information about risk factors for BCC in the general population, see Skin Cancer Prevention.
Sun exposure
Sun exposure is the major known environmental factor associated with the development of skin cancer of all types. There are different patterns of sun exposure associated with each major type of skin cancer (BCC, squamous cell carcinoma [SCC], and melanoma). For more information, see Skin Cancer Prevention.
Pigmentary characteristics
The high-risk phenotype consists of individuals with the following physical characteristics:
- Fair skin that sunburns easily.
- Lightly pigmented irides (blue and green eye color).
- Presence of freckles in sun-exposed skin.
- Poor ability to tan.
- Blond or red hair color.
Specifically, people with more highly pigmented skin demonstrate lower incidence of BCC than do people with lighter pigmented skin. Individuals with Fitzpatrick type I or II skin (lighter skin) were shown to have a twofold increased risk of BCC in a small case-control study. Blond or red hair color was associated with increased risk of BCC in two large cohorts: the Nurses’ Health Study and the Health Professionals’ Follow-Up Study. In women from the Nurses’ Health Study, there was an increased risk of BCC in women with red hair relative to those with light brown hair (adjusted relative risk [RR], 1.30; 95% confidence interval [CI], 1.20–1.40). In men from the Health Professionals Follow-Up Study, the risk of BCC associated with red hair was not as large (RR, 1.17; 95% CI, 1.02–1.34) and was not significant after adjustment for melanoma family history and sunburn history. Risk associated with blond hair was also increased for both men and women (RR, pooled analysis, 1.09; 95% CI, 1.02–1.18), and dark brown hair was protective against BCC (RR, pooled analysis, 0.89; 95% CI, 0.87–0.92). For more information, see the section onPigmentary characteristics in the Melanoma section.
Family history
Individuals with BCCs and/or SCCs report a higher frequency of these cancers in their family members than do controls. The importance of this finding is unclear. Apart from defined genetic disorders with an increased risk of BCC, a positive family history of any skin cancer is a strong predictor of the development of BCC. Data from the Nurses’ Health Study and the Health Professionals Follow-Up Study indicate that the family history of melanoma in a first-degree relative (FDR) is associated with an increased risk of BCC in both men and women (RR, 1.31; 95% CI, 1.25–1.37; P< .0001). A family history of melanoma in the same cohorts, plus the Nurses’ Health Study 2, showed a similar increased risk (hazard ratio [HR], 1.27; 95% CI, 1.12–1.44). A study of 376 early-onset BCC cases and 383 controls found that a family history of any type of skin cancer increased the risk of early-onset BCC (odds ratio [OR], 2.49; 95% CI, 1.80–3.45). This risk increased when an FDR was diagnosed with skin cancer before age 50 years (OR, 4.79; 95% CI, 2.90–7.90). Individuals who had a family history of both melanoma and nonmelanoma skin cancer (NMSC) had the highest risk (OR, 3.65; 95% CI, 1.79–7.47).
A study on the heritability of cancer among 80,309 monozygotic and 123,382 dizygotic twins showed that NMSCs have a heritability of 43% (95% CI, 26%–59%), suggesting that almost half of the risk of NMSC is caused by inherited factors. Additionally, the cumulative risk of NMSC was 1.9-fold higher for monozygotic than for dizygotic twins (95% CI, 1.8–2.0).
Previous personal history of BCC or SCC
A personal history of BCC or SCC is strongly associated with subsequent BCC or SCC. There is an approximate 20% increased risk of a subsequent lesion within the first year after a skin cancer has been diagnosed. The mean age of occurrence for these cancers is the mid-60s. In addition, several studies have found that individuals with a history of BCC or SCC have an increased risk (range, 9%–61%) of a subsequent diagnosis of a noncutaneous cancer; however, other studies have contradicted this finding. In the absence of other risk factors or evidence of a defined cancer susceptibility syndrome, as discussed below, skin cancer patients are encouraged to follow screening recommendations for the general population for sites other than the skin.
Major Genes for Basal Cell Carcinoma
PTCH1
Inherited pathogenic variants in the gene coding for the transmembrane receptor protein PTCH1, or PTCH, are associated with basal cell nevus syndrome (BCNS), and somatic mutations are associated with sporadic cutaneous BCCs. PTCH1, the human homolog of the Drosophila segment polarity gene patched (ptc), is an integral component of the hedgehog signaling pathway, which has many developmental (appendage development, embryonic segmentation, neural tube differentiation) and regulatory (maintenance of stem cells) roles. For more information, see the Basal cell nevus syndrome section.
In the resting state, the transmembrane receptor protein PTCH1 acts catalytically to suppress the seven-transmembrane protein Smoothened (Smo), preventing further downstream signal transduction. Binding of the hedgehog ligand to PTCH1 releases inhibition of Smo, with resultant activation of transcription factors (GLI1, GLI2), cell proliferation genes (cyclin D, cyclin E, myc), and regulators of angiogenesis. Thus, the balance of PTCH1 (inhibition) and Smo (activation) manages the essential regulatory downstream hedgehog signal transduction pathway. Loss-of-function pathogenic variants of PTCH1 or gain-of-function variants of Smo tip this balance toward activation, a key event in potential neoplastic transformation.
Demonstration of allelic loss on chromosome 9q22 in both sporadic and familial BCCs suggested the potential presence of an associated tumor suppressor gene. Further investigation identified a pathogenic variant in PTCH1 that localized to the area of allelic loss. Up to 30% of sporadic BCCs demonstrate PTCH1 pathogenic variants. In addition to BCC, medulloblastoma and rhabdomyosarcoma, along with other tumors, have been associated with PTCH1 pathogenic variants. All three malignancies are associated with BCNS, and most people with clinical features of BCNS demonstrate germlinePTCH1 pathogenic variants, predominantly truncation in type.
PTCH2
Truncating pathogenic variants in PTCH2, a homolog of PTCH1 mapping to chromosome 1p32.1-32.3, have been seen in both BCC and medulloblastoma. PTCH2 displays 57% homology to PTCH1. While the exact role of PTCH2 remains unclear, there is evidence to support its involvement in the hedgehog signaling pathway. However, the role of this gene in BCNS has been contested.
Putative Genes for Basal Cell Carcinoma
BRCA1-associated protein 1 (BAP1)
Pathogenic variants in the BAP1 gene are associated with an increased risk of a variety of cancers, including cutaneous melanoma and uveal melanoma. Although the BCC penetrance in individuals with pathogenic variants in BAP1 is not known, there are several BAP1 families that report diagnoses of BCC. In one study, pathogenic variant carriers from four families reported diagnoses of BCC. Tumor evaluation of BAP1 showed loss of BAP1 protein expression by immunohistochemistry in BCCs of two germline BAP1 pathogenic variant carriers but not in 53 sporadic BCCs. A second report noted that four individuals from families with BAP1 germline pathogenic variants were diagnosed with a total of 19 BCCs. Complete loss of BAP1 nuclear expression was observed in 17 of 19 BCCs from these individuals but none of 22 control BCC specimens. Loss of BAP1 nuclear expression was also reported in a series of 7 BCCs from individuals with loss of function BAP1 variants, but only in 1 of 31 sporadic BCCs. For more information, see the section on BAP1 in the Melanoma section.
MC1R
A meta-analysis showed that the more MC1R pathogenic variants an individual carried, the higher his/her risk was to develop SCC and BCC. Individuals with two or more MC1R pathogenic variants had a summary OR of 2.48 (95% CI, 1.96–3.15) for BCC and a summary OR of 2.80 (95% CI, 1.71–4.57) for SCC; these risks increased when individuals had red hair. A study of individuals diagnosed with BCC before age 40 years also found a stronger association between BCC and MC1R pathogenic variants in those with phenotypic characteristics that are not traditionally considered high risk. For more information, see the section on MC1R in the Melanoma section.
Syndromes Associated With a Predisposition to Basal Cell Carcinoma
Basal cell nevus syndrome
BCNS, also known as Gorlin Syndrome, Gorlin-Goltz syndrome, and nevoid BCC syndrome, is an autosomal dominant disorder with an estimated prevalence of 1 in 57,000 individuals. The syndrome is notable for complete penetrance and high levels of variable expressivity, as evidenced by evaluation of individuals with identical genotypes but widely varying phenotypes. The clinical features of BCNS differ more among families than within families. BCNS is primarily associated with germline pathogenic variants in PTCH1, but families with this phenotype have also been associated with alterations in PTCH2 and SUFU.
As detailed above, PTCH1 provides both developmental and regulatory guidance; spontaneous or inherited germline pathogenic variants of PTCH1 in BCNS may result in a wide spectrum of potentially diagnostic physical findings. The BCNS pathogenic variant has been localized to chromosome 9q22.3-q31, with a maximum logarithm of the odd (LOD) score of 3.597 and 6.457 at markers D9S12 and D9S53. The resulting haploinsufficiency of PTCH1 in BCNS has been associated with structural anomalies such as odontogenic keratocysts, with evaluation of the cyst lining revealing loss of heterozygosity (LOH) for PTCH1. The development of BCC and other BCNS-associated malignancies is thought to arise from the classic two-hit suppressor gene model: baseline heterozygosity secondary to germline PTCH1 pathogenic variant as the first hit, with the second hit due to mutagen exposure such as UV or ionizing radiation. However, haploinsufficiency or dominant negative isoforms have also been implicated for the inactivation of PTCH1.
The diagnosis of BCNS is typically based upon characteristic clinical and radiological examination findings. Several sets of clinical diagnostic criteria for BCNS are in use (for more information, see Table 1). Although each set of criteria has advantages and disadvantages, none of the sets have a clearly superior balance of sensitivity and specificity for identifying carriers of pathogenic variants. The BCNS Colloquium Group proposed criteria in 2011 that required 1 major criterion with molecular diagnosis, two major criteria without molecular diagnosis, or one major and two minor criteria without molecular diagnosis. PTCH1 pathogenic variants are found in 60% to 85% of patients who meet clinical criteria. Most notably, BCNS is associated with the formation of both benign and malignant neoplasms. The strongest benign neoplasm association is with ovarian fibromas, diagnosed in 14% to 24% of females affected by BCNS. BCNS-associated ovarian fibromas are more likely to be bilateral and calcified than sporadic ovarian fibromas. Ameloblastomas, aggressive tumors of the odontogenic epithelium in the jaw, have also been proposed as a diagnostic criterion for BCNS, but most groups do not include it at this time.
Other associated benign neoplasms include gastric hamartomatous polyps, congenital pulmonary cysts, cardiac fibromas, meningiomas, craniopharyngiomas, fetal rhabdomyomas, leiomyomas, mesenchymomas, basaloid follicular hamartomas, and nasal dermoid tumors. Development of meningiomas and ependymomas occurring postradiation therapy has been documented in the general pediatric population; radiation therapy for syndrome-associated intracranial processes may be partially responsible for a subset of these benign tumors in individuals with BCNS. In addition, radiation therapy of malignant medulloblastomas in the BCNS population may result in many cutaneous BCCs in the radiation ports. Similarly, treatment of BCC of the skin with radiation therapy may result in induction of large numbers of additional BCCs.
The diagnostic criteria for BCNS are described in Table 1 below.
| Evans et al. 1993 | Kimonis et al. 1997 | Veenstra-Knol et al. 2005 | BCNS Colloquium Group 2011b |
|---|---|---|---|
| BCC = basal cell carcinoma. | |||
| aTwo major criteria or one major and two minor criteria needed to meet the requirements for a BCNS diagnosis. | |||
| bDiagnosis is based on one major criterion with molecular diagnosis, two major criteria without molecular diagnosis, or one major and two minor criteria without molecular diagnosis. | |||
| Major Criteriaa | |||
| >2 BCCs or 1 BCC diagnosed before age 30 y or >10 basal cell nevi | >2 BCCs or 1 BCC diagnosed before age 20 y | >2 BCCs or 1 BCC diagnosed before age 20 y | BCC before age 20 y or excessive number of BCCs out of proportion with previous skin exposure and skin type |
| Histologically proven odontogenic keratocyst of jaw or polyostotic bone cyst | Histologically proven odontogenic keratocyst of jaw | Histologically proven odontogenic keratocyst of jaw | Odontogenic keratocyst of jaw before age 20 y |
| ≥3 palmar or plantar pits | ≥3 palmar or plantar pits | ≥3 palmar or plantar pits | Palmar or plantar pitting |
| Ectopic calcifications, lamellar or early (diagnosed before age 20 y) falx calcifications in brain | Bilamellar calcification of falx cerebri in brain | Ectopic calcification (lamellar or early falx cerebri) in brain | Lamellar calcification of falx cerebri in brain |
| Family history of BCNS | First-degree relative with BCNS | Family history of BCNS | First-degree relative with BCNS |
| (Rib abnormalities listed as minor criterion; see below) | Bifid, fused, or splayed ribs | Bifid, fused, or splayed ribs | (Rib abnormalities listed as minor criterion; see below) |
| (Medulloblastoma listed as minor criterion; see below) | (Medulloblastoma listed as minor criterion; see below) | (Medulloblastoma listed as minor criterion; see below) | Medulloblastoma (usually desmoplastic) |
| Minor Criteria | |||
| Occipital-frontal circumference >97th percentile and frontal bossing | Macrocephaly (adjusted for height) | Macrocephaly (>97th percentile) | Macrocephaly |
| Congenital skeletal abnormalities: bifid, fused, splayed, or missing rib or bifid, wedged, or fused vertebrae | Bridging of sella turcica, vertebral abnormalities (hemivertebrae, fusion or elongation of vertebral bodies), modeling defects of the hands and feet, or flame-shaped lucencies of hands and feet on x-ray | Bridging of sella turcica, vertebral abnormalities (hemivertebrae, fusion or elongation of vertebral bodies), modeling defects of the hands and feet | Skeletal malformations (vertebral, short 4th metacarpals, postaxial polydactyly) |
| (Rib abnormalities listed as major criterion; see above) | (Rib abnormalities listed as major criterion; see above) | Rib abnormalities | |
| Cardiac or ovarian fibroma | Ovarian fibroma | Cardiac or ovarian fibroma | Cardiac or ovarian fibroma |
| Medulloblastoma | Medulloblastoma | Medulloblastoma | (Medulloblastoma listed as major criterion; see above) |
| Congenital malformation: cleft lip and/or palate, polydactyly, cataract, coloboma, microphthalmia | Cleft lip or palate, frontal bossing, moderate or severe hypotelorism | Cleft lip and/or palate, polydactyly | Cleft lip or palate |
| Sprengel deformity, marked pectus deformity, marked syndactyly | Sprengel deformity, marked pectus deformity, marked syndactyly | ||
| Lymphomesenteric cysts | Lymphomesenteric cysts | ||
| Eye anomaly: cataract, coloboma, microphthalmia | Ocular abnormalities (strabismus, hypertelorism, Congenital cataracts, coloboma) | ||
Of greatest concern with BCNS are associated malignant neoplasms, the most common of which is BCC. BCC in individuals with BCNS may appear during childhood as small acrochordon-like lesions, while larger lesions demonstrate more classic cutaneous features. Nonpigmented BCCs are more common than pigmented lesions. The age at first BCC diagnosis associated with BCNS ranges from 3 to 53 years, with a mean age of 21.4 years; the vast majority of individuals are diagnosed with their first BCC before age 20 years. Most BCCs are located on sun-exposed sites, but individuals with greater than 100 BCCs have a more uniform distribution of BCCs over the body. Case series have suggested that up to 1 in 200 individuals with BCC demonstrate findings supportive of a diagnosis of BCNS. BCNS has rarely been reported in individuals with darker skin pigmentation; however, significantly fewer BCCs are found in individuals of African or Mediterranean ancestry. Despite the rarity of BCC in this population, reported cases document full expression of the noncutaneous manifestations of BCNS. However, in individuals of African ancestry who have received radiation therapy, significant basal cell tumor burden has been reported within the radiation port distribution. Thus, cutaneous pigmentation may protect against the mutagenic effects of UV but not against ionizing radiation.
Variants in other genes associated with an increased risk of BCC in the general population appear to modify the age of BCC onset in individuals with BCNS. A study of 125 individuals with BCNS found that a variant in MC1R (Arg151Cys) was associated with an early median age of onset of 27 years (95% CI, 20–34), compared with individuals who did not carry the risk allele and had a median age of BCC of 34 years (95% CI, 30–40) (HR, 1.64; 95% CI, 1.04–2.58, P = .034). A variant in the TERT-CLPTM1L gene showed a similar effect, with individuals with the risk allele having a median age of BCC of 31 years (95% CI, 28–37) relative to a median onset of 41 years (95% CI, 32–48) in individuals who did not carry a risk allele (HR, 1.44; 95% CI, 1.08–1.93, P = .014).
Many other malignancies have been associated with BCNS. Medulloblastoma carries the strongest association with BCNS and is diagnosed in 1% to 5% of BCNS cases. While BCNS-associated medulloblastoma is typically diagnosed between ages 2 and 3 years, sporadic medulloblastoma is usually diagnosed later in childhood, between the ages of 6 and 10 years. A desmoplastic phenotype occurring around age 2 years is very strongly associated with BCNS and carries a more favorable prognosis than sporadic classic medulloblastoma. Up to three times more males than females with BCNS are diagnosed with medulloblastoma. As with other malignancies, treatment of medulloblastoma with ionizing radiation has resulted in numerous BCCs within the radiation field. Other reported malignancies include ovarian carcinoma, ovarian fibrosarcoma, astrocytoma, melanoma, Hodgkin disease, rhabdomyosarcoma, and undifferentiated sinonasal carcinoma.
Odontogenic keratocysts–or keratocystic odontogenic tumors (KCOTs), as renamed by the World Health Organization working group–are one of the major features of BCNS. Demonstration of clonal LOH of common tumor suppressor genes, including PTCH1, supports the transition of terminology to reflect a neoplastic process. Less than one-half of KCOTs from individuals with BCNS show LOH of PTCH1. The tumors are lined with a thin squamous epithelium and a thin corrugated layer of parakeratin. Increased mitotic activity in the tumor epithelium and potential budding of the basal layer with formation of daughter cysts within the tumor wall may be responsible for the high rates of recurrence post simple enucleation. In a recent case series of 183 consecutively excised KCOTs, 6% of individuals demonstrated an association with BCNS. A study that analyzed the rate of PTCH1 pathogenic variants in BCNS-associated KCOTs found that 11 of 17 individuals carried a germline PTCH1 pathogenic variant and an additional 3 individuals had somatic mutations in this gene. Individuals with germline PTCH1 pathogenic variants had an early age of KCOT presentation. KCOTs occur in 65% to 100% of individuals with BCNS, with higher rates of occurrence in young females.
Palmoplantar pits are another major finding in BCC and occur in 70% to 80% of individuals with BCNS. When these pits occur together with early-onset BCC and/or KCOTs, they are considered diagnostic for BCNS.
Several characteristic radiological findings have been associated with BCNS, including lamellar calcification of falx cerebri in the brain; fused, splayed or bifid ribs; and flame-shaped lucencies or pseudocystic bone lesions of the phalanges, carpal, tarsal, long bones, pelvis, and calvaria by diagnostic x-ray imaging. Imaging for rib abnormalities may be useful when establishing a BCNS diagnosis in young children, who may not have fully manifested BCNS features that are detected during physical examination. The presence of skeletal abnormalities appears to affect BCC number and severity in affected individuals. In one study, individuals with BCNS and skeletal findings had a mean of 120 more BCCs than those who had BCNS without skeletal findings. Individuals with two or more skeletal changes also had an increased risk of advanced or metastatic BCC (OR, 2.45; 95% CI, 1.01–5.91).
Table 2 summarizes the frequency and median age of onset of nonmalignant findings associated with BCNS.
| Finding | Frequency (%) | Median Age of Onset |
|---|---|---|
| Adapted from a report by Kimonis et al. about 105 individuals with BCNS seen at the National Institutes of Health between 1985 and 1997. | ||
| Palmar/plantar pits | 87 | Usually by age 10 y |
| Keratogenic jaw cysts | 74 | Usually by age 20 y |
| Bridged sella | 68 | Congenital |
| Calcification of falx cerebri | 65 | Usually by age 40 y |
| Macrocephaly | 50 | Congenital |
| Hypertelorism | 42 | Congenital |
| Osseous lucencies in the hands | 30 | Congenital |
| Frontal bossing | 27 | Congenital |
| Bifid ribs | 26 | Congenital |
| Calcification of tentorium cerebelli | 20 | Not reported |
| Ovarian fibromas | 17 | 30 y |
| Hemivertebra | 15 | Congenital |
| Pectus deformity | 11 | Congenital |
| Fusion of vertebral bodies | 10 | Congenital |
| Cleft lip/palate | 3 | Congenital |
Individuals with PTCH2 pathogenic variants may have a milder phenotype of BCNS than those with PTCH1 variants. Characteristic features such as palmar/plantar pits, macrocephaly, falx calcification, hypertelorism, and coarse face may be absent in these individuals.
A 9p22.3 microdeletion syndrome that includes the PTCH1locus has been described in ten children. All patients had facial features typical of BCNS, including a broad forehead, but they had other features variably including craniosynostosis, hydrocephalus, macrosomia, and developmental delay. At the time of the report, none had basal cell skin cancer. On the basis of their hemizygosity of the PTCH1 gene, these patients are presumably at an increased risk of basal cell skin cancer.
Germline pathogenic variants in SUFU, a major negative regulator of the hedgehog pathway, have been identified in a small number of individuals with a clinical phenotype resembling that of BCNS. These pathogenic variants were first identified in individuals with childhood medulloblastoma, and the incidence of medulloblastoma appears to be much higher in individuals with BCNS associated with SUFU pathogenic variants than in those with PTCH1 variants. SUFU pathogenic variants may also be associated with an increased predisposition to meningioma. Conversely, odontogenic jaw keratocysts appear less frequently in this population. Some clinical laboratories offer genetic testing for SUFU pathogenic variants for individuals with BCNS.
DNA repair genes
In addition to pathogenic variants in genes primarily associated with BCC, other cancer-associated genes may confer an increased risk for BCC. A study of 61 individuals with a high number of BCCs (mean, 11 BCCs; range, 6–65) underwent genetic testing for 29 high-penetrance cancer susceptibility genes. Thirteen pathogenic variants were found in 12 of 61 individuals (19.7%). This was higher than expected compared with individuals in the Exome Aggregation Consortium (ExAC) database (3%). All of the genes with pathogenic variants were involved in DNA repair, suggesting that defects in DNA repair pathways may increase the risk of BCC. Of these 61 individuals, 21 (34.4%) had a previous diagnosis of another cancer including melanoma, breast, colon, and prostate cancers.
Xeroderma pigmentosum
Xeroderma pigmentosum (XP) is a hereditary disorder of nucleotide excision repair that results in cutaneous malignancies in the first decade of life. Affected individuals have an increased sensitivity to sunlight, resulting in a markedly increased risk of SCCs, BCCs, and melanomas. For more information, see the section on Xeroderma pigmentosum in the Squamous Cell Carcinoma section.
Rare syndromes
Rombo syndrome
Rombo syndrome is a very rare genodermatosis or genetic disorder associated with BCC. It is thought to have an autosomal dominant inheritance pattern, and it has been outlined in three case series in the literature. The cutaneous examination is within normal limits until age 7 to 10 years, with the development of distinctive cyanoticerythema of the lips, hands, and feet and early atrophoderma vermiculatum of the cheeks, with variable involvement of the elbows and dorsal hands and feet. Development of BCC occurs in the fourth decade. A distinctive grainy texture to the skin, secondary to interspersed small, yellowish, follicular-based papules and follicular atrophy, has been described. Missing, irregularly distributed, and/or misdirected eyelashes and eyebrows are another associated finding. The genetic basis of Rombo syndrome is not known.
Bazex-Dupré-Christol syndrome
Bazex-Dupré-Christol syndrome, another rare genodermatosis associated with development of BCC, has more thorough documentation in the literature than Rombo syndrome. Inheritance is accomplished in an X-linked dominant fashion, with no reported male-to-male transmission. Regional assignment of the locus of interest to chromosome Xq24-q27 is associated with a maximum LOD score of 5.26 with the DXS1192 locus. Further work has narrowed the potential location to an 11.4-Mb interval on chromosome Xq25-27; however, the causative gene remains unknown.
Characteristic physical findings include hypotrichosis, hypohidrosis, milia, follicular atrophoderma of the cheeks, and multiple BCC, which manifest in the late second decade to early third decade. Documented hair changes with Bazex-Dupré-Christol syndrome include reduced density of scalp and body hair, decreased melanization, a twisted/flattened appearance of the hair shaft on electron microscopy, and increased hair shaft diameter on polarizing light microscopy. The milia, which may be quite distinctive in childhood, have been reported to regress or diminish substantially at puberty. Other reported findings in association with this syndrome include trichoepitheliomas; hidradenitis suppurativa; hypoplastic alae; and a prominent columella, the fleshy terminal portion of the nasal septum.
Epidermolysis bullosa simplex
A rare, severe subtype of epidermolysis bullosa simplex (EBS), previously known as Dowling-Meara (EBS-DM), is primarily inherited in an autosomal dominant fashion and is associated with pathogenic variants in either keratin-5 (KRT5) or keratin-14 (KRT14). This severe subtype of EBS occasionally results in mortality in early childhood. It has an estimated prevalence of 0.02 per million individuals in the United States and an incidence of 1.16 per million live births. One report cites an incidence of BCC of 44% by age 55 years in this population. Individuals who inherit two EBS pathogenic variants may present with a more severe phenotype. Other less phenotypically severe subtypes of EBS can also be caused by pathogenic variants in either KRT5 or KRT14. Approximately 75% of individuals with a clinical diagnosis of EBS (regardless of subtype) have KRT5 or KRT14 pathogenic variants.
Characteristics of hereditary syndromes associated with a predisposition to BCC are described in Table 3 below.
| Syndrome | Inheritance | Gene or Chromosomal Loci | Clinical Findings | |
|---|---|---|---|---|
| AD = autosomal dominant; AR = autosomal recessive; SCC = squamous cell carcinoma; XD = X-linked dominant. | ||||
| Basal cell nevus syndrome, Gorlin syndrome | AD | PTCH1, PTCH2, SUFU | BCC (before age 20 y) | |
| Rombo syndrome | AD | Unknown | Milia, atrophoderma vermiculatum, acrocyanosis, trichoepitheliomas, and BCC (age 30–40 y) | |
| Bazex-Dupré-Christol syndrome | XD > AD | Xq24-27 | Hypotrichosis (variable), hypohidrosis, milia, follicular atrophoderma (dorsal hands), and multiple BCCs (aged teens to early 20s) | |
| Brooke-Spiegler syndrome | AD | CYLD | Cylindroma (forehead, scalp, trunk, and pubic area), trichoepithelioma (around nose), spiradenoma, and BCC | |
| Multiple hereditary infundibulocystic BCC | AD | Unknown | Multiple BCC (infundibulocystic type) | |
| Schopf-Schultz-Passarge syndrome | AR > AD | Unknown | Ectodermal dysplasia (hypotrichosis, hypodontia, and nail dystrophy [anonychia and trachyonychia]), hidrocystomas of eyelids, palmoplantar keratosis and hyperhidrosis, and BCC | |
| Xeroderma pigmentosum | AR | XPA, XPB/ERCC3, XPC, XPD/ERCC2, XPE/DDB2, XPF/ERCC4, XPG/ERCC5 | SCC, BCC, melanoma, severe sun sensitivity, ophthalmologic and neurologic abnormalities | |
| Xeroderma pigmentosum variant | AR | POLH | SCC, BCC, melanoma, severe sun sensitivity, ophthalmologic abnormalities | |
For more information, see the Brooke-Spiegler Syndrome, Multiple Familial Trichoepithelioma, and Familial Cylindromatosis section.
Interventions
Screening
As detailed further below, the U.S. Preventive Services Task Force does not recommend regular screening for the early detection of any cutaneous malignancies, including BCC. However, once a BCC is detected on the skin of an individual, the National Comprehensive Cancer Network recommends that he/she have a complete skin examination biannually or annually for the first 5 years after the BCC is detected. After 5 years, skin examinations are recommended at least once a year for life.
Table 4 summarizes available clinical practice guidelines for the surveillance of individuals with BCNS.
| MRI = magnetic resonance imaging. | |
| Adapted from Bree et al. and Foulkes et al. | |
| For Adults: | |
| • MRI of brain (baseline) | |
| • Skin examination every 4 months | |
| • Panorex of jaw every year | |
| • Neurological evaluation (if previous medulloblastoma) | |
| • Pelvic ultrasound (baseline) | |
| • Gynecologic examination every year | |
| • Nutritional assessment | |
| • Fetal assessment for hydrocephalus, macrocephaly, and cardiac fibromas in pregnancy | |
| • Minimization of diagnostic radiation exposure when feasible | |
| For Children: | |
| • MRI of brain (annually until age 8 years) | |
| • Low risk (PTCH1): No radiographic screening unless concerning neurological exam, head circumference change, or other unusual signs/symptoms | |
| • High risk (SUFU): Brain MRI every 4 months through age 3 years, then every 6 months until age 5 years | |
| • Cardiac ultrasound (baseline) | |
| • Dermatologic examination (baseline) | |
| • Annual by age 10 years, increased frequency after first basal cell carcinoma is diagnosed | |
| • Panorex of jaw (baseline, then annually if no cysts apparent; after the first cyst is diagnosed, every 6 months until age 21 years or until no cysts are noted for two years) | |
| • Beginning at age 8 years, then every 12–18 months | |
| • Some dermatologists recommend waiting until symptomatic to begin Panorex in order to limit radiation exposure | |
| • Spine film at age 1 year or time of diagnosis (if abnormal, follow scoliosis protocol) | |
| • Pelvic ultrasound at menarche or age 18 years | |
| • Hearing, speech, and ophthalmologic evaluation | |
| • Minimization of diagnostic radiation exposure when feasible | |
Level of evidence: 5
Primary prevention
Avoidance of excessive cumulative and sporadic sun exposure is important in reducing the risk of BCC, along with other cutaneous malignancies. Scheduling activities outside of the peak hours of UV radiation, utilizing sun-protective clothing and hats, using sunscreen liberally, and strictly avoiding tanning beds are all reasonable steps towards minimizing future risk of skin cancer. For patients with particular genetic susceptibility (such as BCNS), avoidance or minimization of ionizing radiation is essential to reducing future tumor burden.
Level of evidence: 2aii
Chemoprevention
The role of various systemic retinoids, including isotretinoin and acitretin, has been explored in the chemoprevention and treatment of multiple BCCs, particularly in BCNS patients. In one study of isotretinoin use in 12 patients with multiple BCCs, including 5 patients with BCNS, tumor regression was noted, with decreasing efficacy as the tumor diameter increased. However, the results were insufficient to recommend use of systemic retinoids for treatment of BCC. Three additional patients, including one with BCNS, were followed long-term for evaluation of chemoprevention with isotretinoin, demonstrating significant decrease in the number of tumors per year during treatment. Although the rate of tumor development tends to increase sharply upon discontinuation of systemic retinoid therapy, in some patients the rate remains lower than their pretreatment rate, allowing better management and control of their cutaneous malignancies. In summary, the use of systemic retinoids for chemoprevention of BCC is reasonable in high-risk patients, including patients with xeroderma pigmentosum. For more information, see the Squamous Cell Carcinoma section.
A patient’s cumulative and evolving tumor load should be evaluated carefully in light of the potential long-term use of a medication class with cumulative and idiosyncratic side effects. Given the possible side-effect profile, systemic retinoid use is best managed by a practitioner with particular expertise and comfort with the medication class. However, for all potentially childbearing women, strict avoidance of pregnancy during the systemic retinoid course—and for 1 month after completion of isotretinoin and 3 years after completion of acitretin—is essential to avoid potentially fatal and devastating fetal malformations. In the United States, isotretinoin can only be prescribed through the U.S. Food and Drug Administration (FDA)-mandated iPledge program.
Level of evidence (retinoids): 2aii
In a phase II study of 41 patients with BCNS, vismodegib (an inhibitor of the hedgehog pathway) has been shown to reduce the per-patient annual rate of new BCCs requiring surgery. Existing BCCs also regressed for these patients during daily treatment with 150 mg of oral vismodegib. While patients treated had visible regression of their tumors, biopsy demonstrated residual microscopic malignancies at the site, and tumors progressed after the discontinuation of the therapy. Adverse effects included taste disturbance, muscle cramps, hair loss, and weight loss and led to discontinuation of the medication in 54% of subjects. A subsequent, open-label, phase II study included 37 patients from the same cohort who continued vismodegib for up to a total of 36 months. Patients treated with vismodegib had a lower mean incidence of new, surgically eligible BCCs than did placebo-treated patients (P< .0001). However, only 17% of patients tolerated continuous vismodegib for the full 36 months. Tumors reappeared after treatment was stopped, but patients who resumed treatment again experienced tumor response. The duration of benefit after stopping vismodegib appeared to be proportional to the duration and compliance of taking the drug during treatment. Intermittent dosing schedules of vismodegib (8 weeks on/8 weeks off after an initial schedule of daily dosing for 24 weeks or 12 weeks on/8 weeks off) have also been shown to be effective in the reduction of BCCs in the BCNS population, although there has been no direct comparison between continuous dosing and intermittent dosing schedules. On the basis of the side-effect profile and rate of disease recurrence after discontinuation of the medication, additional study regarding optimal dosing of vismodegib is ongoing.
Level of evidence (vismodegib): 1aii
A phase III, double-blind, placebo-controlled clinical trial evaluated the effects of oral nicotinamide (vitamin B3) in 386 individuals with a history of at least two keratinocyte carcinomas (BCC or SCC) within 5 years before study enrollment. After 12 months of treatment, those taking nicotinamide 500 mg twice daily had a 20% reduction in the incidence of new BCCs (95% CI, 6%–39%; P = .12). The rate of new keratinocyte carcinomas was 23% lower in the nicotinamide group (95% CI, 4%–38%; P = .02) than in the placebo group. No clinically significant differences in adverse events were observed between the two groups, and there was no evidence of benefit after discontinuation of nicotinamide. Of note, this study was not conducted in a population with an identified genetic predisposition to BCC.
Level of evidence (nicotinamide): 1aii
Treatment
Treatment of individual BCCs in BCNS is generally the same as for sporadic basal cell cancers. Due to the large number of lesions on some patients, this can present a surgical challenge. Field therapy with imiquimod or photodynamic therapy are attractive options, as they can treat multiple tumors simultaneously. However, given the radiosensitivity of patients with BCNS, radiation as a therapeutic option for large tumors should be avoided. There are no randomized trials, but the isolated case reports suggest that field therapy has similar results as in sporadic basal cell cancer, with higher success rates for superficial cancers than for nodular cancers.
Consensus guidelines for the use of methylaminolevulinate photodynamic therapy in BCNS recommend that this modality may best be used for superficial BCC of all sizes and for nodular BCC less than 2 mm thick. Monthly therapy with photodynamic therapy may be considered for these patients as clinically indicated.
Level of evidence (imiquimod and photodynamic therapy): 4
Topical treatment with LDE225, a Smoothened agonist, has also been investigated for the treatment of BCC in a small number of patients with BCNS with promising results; however, this medication is not approved in this formulation by the FDA.
Level of evidence (LDE225): 1
In addition to its effects on the prevention of BCCs in patients with BCNS, vismodegib may also have a palliative effect on KCOTs found in this population. An initial report indicated that the use of GDC-0449, the hedgehog pathway inhibitor now known as vismodegib, resulted in resolution of KCOTs in one patient with BCNS. Another small study found that four of six patients who took 150 mg of vismodegib daily had a reduction in the size of KCOTs. None of the six patients in this study had new KCOTs or an increase in the size of existing KCOTs while being treated, and one patient had a sustained response that lasted 9 months after treatment was discontinued.
Level of evidence (vismodegib): 3diii
References
- Miller DL, Weinstock MA: Nonmelanoma skin cancer in the United States: incidence. J Am Acad Dermatol 30 (5 Pt 1): 774-8, 1994.
- Gon A, Minelli L: Risk factors for basal cell carcinoma in a southern Brazilian population: a case-control study. Int J Dermatol 50 (10): 1286-90, 2011.
- Wu S, Han J, Li WQ, et al.: Basal-cell carcinoma incidence and associated risk factors in U.S. women and men. Am J Epidemiol 178 (6): 890-7, 2013.
- Wei EX, Li X, Nan H: Having a first-degree relative with melanoma increases lifetime risk of melanoma, squamous cell carcinoma, and basal cell carcinoma. J Am Acad Dermatol 81 (2): 489-499, 2019.
- Berlin NL, Cartmel B, Leffell DJ, et al.: Family history of skin cancer is associated with early-onset basal cell carcinoma independent of MC1R genotype. Cancer Epidemiol 39 (6): 1078-83, 2015.
- Mucci LA, Hjelmborg JB, Harris JR, et al.: Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 315 (1): 68-76, 2016.
- Epstein E: Value of follow-up after treatment of basal cell carcinoma. Arch Dermatol 108 (6): 798-800, 1973.
- Møller R, Nielsen A, Reymann F: Multiple basal cell carcinoma and internal malignant tumors. Arch Dermatol 111 (5): 584-5, 1975.
- Bergstresser PR, Halprin KM: Multiple sequential skin cancers. The risk of skin cancer in patients with previous skin cancer. Arch Dermatol 111 (8): 995-6, 1975.
- Robinson JK: Risk of developing another basal cell carcinoma. A 5-year prospective study. Cancer 60 (1): 118-20, 1987.
- Greenberg ER, Baron JA, Stukel TA, et al.: A clinical trial of beta carotene to prevent basal-cell and squamous-cell cancers of the skin. The Skin Cancer Prevention Study Group. N Engl J Med 323 (12): 789-95, 1990.
- Karagas MR, Stukel TA, Greenberg ER, et al.: Risk of subsequent basal cell carcinoma and squamous cell carcinoma of the skin among patients with prior skin cancer. Skin Cancer Prevention Study Group. JAMA 267 (24): 3305-10, 1992.
- Cantwell MM, Murray LJ, Catney D, et al.: Second primary cancers in patients with skin cancer: a population-based study in Northern Ireland. Br J Cancer 100 (1): 174-7, 2009.
- Efird JT, Friedman GD, Habel L, et al.: Risk of subsequent cancer following invasive or in situ squamous cell skin cancer. Ann Epidemiol 12 (7): 469-75, 2002.
- Wheless L, Black J, Alberg AJ: Nonmelanoma skin cancer and the risk of second primary cancers: a systematic review. Cancer Epidemiol Biomarkers Prev 19 (7): 1686-95, 2010.
- Frisch M, Hjalgrim H, Olsen JH, et al.: Risk for subsequent cancer after diagnosis of basal-cell carcinoma. A population-based, epidemiologic study. Ann Intern Med 125 (10): 815-21, 1996.
- Cho HG, Kuo KY, Li S, et al.: Frequent basal cell cancer development is a clinical marker for inherited cancer susceptibility. JCI Insight 3 (15): , 2018.
- Small J, Wallace K, Hill EG, et al.: A cohort study of personal and family history of skin cancer in relation to future risk of non-cutaneous malignancies. Cancer Causes Control 30 (11): 1213-1221, 2019.
- Tuohimaa P, Pukkala E, Scélo G, et al.: Does solar exposure, as indicated by the non-melanoma skin cancers, protect from solid cancers: vitamin D as a possible explanation. Eur J Cancer 43 (11): 1701-12, 2007.
- de Vries E, Soerjomataram I, Houterman S, et al.: Decreased risk of prostate cancer after skin cancer diagnosis: a protective role of ultraviolet radiation? Am J Epidemiol 165 (8): 966-72, 2007.
- Grant WB: A meta-analysis of second cancers after a diagnosis of nonmelanoma skin cancer: additional evidence that solar ultraviolet-B irradiance reduces the risk of internal cancers. J Steroid Biochem Mol Biol 103 (3-5): 668-74, 2007.
- Soerjomataram I, Louwman WJ, Lemmens VE, et al.: Are patients with skin cancer at lower risk of developing colorectal or breast cancer? Am J Epidemiol 167 (12): 1421-9, 2008.
- Tabata T, Kornberg TB: Hedgehog is a signaling protein with a key role in patterning Drosophila imaginal discs. Cell 76 (1): 89-102, 1994.
- Lum L, Beachy PA: The Hedgehog response network: sensors, switches, and routers. Science 304 (5678): 1755-9, 2004.
- Tojo M, Kiyosawa H, Iwatsuki K, et al.: Expression of the GLI2 oncogene and its isoforms in human basal cell carcinoma. Br J Dermatol 148 (5): 892-7, 2003.
- Gailani MR, Bale SJ, Leffell DJ, et al.: Developmental defects in Gorlin syndrome related to a putative tumor suppressor gene on chromosome 9. Cell 69 (1): 111-7, 1992.
- Shanley SM, Dawkins H, Wainwright BJ, et al.: Fine deletion mapping on the long arm of chromosome 9 in sporadic and familial basal cell carcinomas. Hum Mol Genet 4 (1): 129-33, 1995.
- Hahn H, Christiansen J, Wicking C, et al.: A mammalian patched homolog is expressed in target tissues of sonic hedgehog and maps to a region associated with developmental abnormalities. J Biol Chem 271 (21): 12125-8, 1996.
- Gailani MR, Ståhle-Bäckdahl M, Leffell DJ, et al.: The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet 14 (1): 78-81, 1996.
- Wicking C, Shanley S, Smyth I, et al.: Most germ-line mutations in the nevoid basal cell carcinoma syndrome lead to a premature termination of the PATCHED protein, and no genotype-phenotype correlations are evident. Am J Hum Genet 60 (1): 21-6, 1997.
- Smyth I, Narang MA, Evans T, et al.: Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene in basal cell carcinoma and medulloblastoma on chromosome 1p32. Hum Mol Genet 8 (2): 291-7, 1999.
- Shakhova O, Leung C, van Montfort E, et al.: Lack of Rb and p53 delays cerebellar development and predisposes to large cell anaplastic medulloblastoma through amplification of N-Myc and Ptch2. Cancer Res 66 (10): 5190-200, 2006.
- Goodrich LV, Johnson RL, Milenkovic L, et al.: Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev 10 (3): 301-12, 1996.
- Rahnama F, Toftgård R, Zaphiropoulos PG: Distinct roles of PTCH2 splice variants in Hedgehog signalling. Biochem J 378 (Pt 2): 325-34, 2004.
- Smith MJ, Evans DG: PTCH2 is not a strong candidate gene for gorlin syndrome predisposition. Fam Cancer 21 (3): 343-346, 2022.
- Wadt KA, Aoude LG, Johansson P, et al.: A recurrent germline BAP1 mutation and extension of the BAP1 tumor predisposition spectrum to include basal cell carcinoma. Clin Genet 88 (3): 267-72, 2015.
- Carbone M, Flores EG, Emi M, et al.: Combined Genetic and Genealogic Studies Uncover a Large BAP1 Cancer Syndrome Kindred Tracing Back Nine Generations to a Common Ancestor from the 1700s. PLoS Genet 11 (12): e1005633, 2015.
- de la Fouchardière A, Cabaret O, Savin L, et al.: Germline BAP1 mutations predispose also to multiple basal cell carcinomas. Clin Genet 88 (3): 273-7, 2015.
- Mochel MC, Piris A, Nose V, et al.: Loss of BAP1 Expression in Basal Cell Carcinomas in Patients With Germline BAP1 Mutations. Am J Clin Pathol 143 (6): 901-4, 2015.
- Tagliabue E, Fargnoli MC, Gandini S, et al.: MC1R gene variants and non-melanoma skin cancer: a pooled-analysis from the M-SKIP project. Br J Cancer 113 (2): 354-63, 2015.
- Ferrucci LM, Cartmel B, Molinaro AM, et al.: Host phenotype characteristics and MC1R in relation to early-onset basal cell carcinoma. J Invest Dermatol 132 (4): 1272-9, 2012.
- Farndon PA, Del Mastro RG, Evans DG, et al.: Location of gene for Gorlin syndrome. Lancet 339 (8793): 581-2, 1992.
- Shimkets R, Gailani MR, Siu VM, et al.: Molecular analysis of chromosome 9q deletions in two Gorlin syndrome patients. Am J Hum Genet 59 (2): 417-22, 1996.
- Bale AE: Variable expressivity of patched mutations in flies and humans. Am J Hum Genet 60 (1): 10-2, 1997.
- Fan Z, Li J, Du J, et al.: A missense mutation in PTCH2 underlies dominantly inherited NBCCS in a Chinese family. J Med Genet 45 (5): 303-8, 2008.
- Smith MJ, Beetz C, Williams SG, et al.: Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol 32 (36): 4155-61, 2014.
- Pastorino L, Ghiorzo P, Nasti S, et al.: Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A 149A (7): 1539-43, 2009.
- Agaram NP, Collins BM, Barnes L, et al.: Molecular analysis to demonstrate that odontogenic keratocysts are neoplastic. Arch Pathol Lab Med 128 (3): 313-7, 2004.
- High A, Zedan W: Basal cell nevus syndrome. Curr Opin Oncol 17 (2): 160-6, 2005.
- Bacanli A, Ciftcioglu MA, Savas B, et al.: Nevoid basal cell carcinoma syndrome associated with unilateral renal agenesis: acceleration of basal cell carcinomas following radiotherapy. J Eur Acad Dermatol Venereol 19 (4): 510-1, 2005.
- Strong LC: Genetic and environmental interactions. Cancer 40 (4 Suppl): 1861-6, 1977.
- Evans DG, Birch JM, Orton CI: Brain tumours and the occurrence of severe invasive basal cell carcinoma in first degree relatives with Gorlin syndrome. Br J Neurosurg 5 (6): 643-6, 1991.
- Levanat S, Gorlin RJ, Fallet S, et al.: A two-hit model for developmental defects in Gorlin syndrome. Nat Genet 12 (1): 85-7, 1996.
- Pan S, Dong Q, Sun LS, et al.: Mechanisms of inactivation of PTCH1 gene in nevoid basal cell carcinoma syndrome: modification of the two-hit hypothesis. Clin Cancer Res 16 (2): 442-50, 2010.
- Evans DG, Ladusans EJ, Rimmer S, et al.: Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet 30 (6): 460-4, 1993.
- Kimonis VE, Goldstein AM, Pastakia B, et al.: Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet 69 (3): 299-308, 1997.
- Veenstra-Knol HE, Scheewe JH, van der Vlist GJ, et al.: Early recognition of basal cell naevus syndrome. Eur J Pediatr 164 (3): 126-30, 2005.
- Bree AF, Shah MR; BCNS Colloquium Group: Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS). Am J Med Genet A 155A (9): 2091-7, 2011.
- Klein RD, Dykas DJ, Bale AE: Clinical testing for the nevoid basal cell carcinoma syndrome in a DNA diagnostic laboratory. Genet Med 7 (9): 611-9, 2005 Nov-Dec.
- Kimonis VE, Mehta SG, Digiovanna JJ, et al.: Radiological features in 82 patients with nevoid basal cell carcinoma (NBCC or Gorlin) syndrome. Genet Med 6 (6): 495-502, 2004 Nov-Dec.
- Shanley S, Ratcliffe J, Hockey A, et al.: Nevoid basal cell carcinoma syndrome: review of 118 affected individuals. Am J Med Genet 50 (3): 282-90, 1994.
- Scully RE, Galdabini JJ, McNeely BU: Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 14-1976. N Engl J Med 294 (14): 772-7, 1976.
- Ponti G, Pastorino L, Pollio A, et al.: Ameloblastoma: a neglected criterion for nevoid basal cell carcinoma (Gorlin) syndrome. Fam Cancer 11 (3): 411-8, 2012.
- Schwartz RA: Basal-cell-nevus syndrome and gastrointestinal polyposis. N Engl J Med 299 (1): 49, 1978.
- Totten JR: The multiple nevoid basal cell carcinoma syndrome. Report of its occurrence in four generations of a family. Cancer 46 (6): 1456-62, 1980.
- Jones KL, Wolf PL, Jensen P, et al.: The Gorlin syndrome: a genetically determined disorder associated with cardiac tumor. Am Heart J 111 (5): 1013-5, 1986.
- Gorlin RJ: Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore) 66 (2): 98-113, 1987.
- Mortimer PS, Geaney DP, Liddell K, et al.: Basal cell naevus syndrome and intracranial meningioma. J Neurol Neurosurg Psychiatry 47 (2): 210-2, 1984.
- Kijima C, Miyashita T, Suzuki M, et al.: Two cases of nevoid basal cell carcinoma syndrome associated with meningioma caused by a PTCH1 or SUFU germline mutation. Fam Cancer 11 (4): 565-70, 2012.
- Tamoney HJ: Basal cell nevoid syndrome. Am Surg 35 (4): 279-83, 1969.
- DiSanto S, Abt AB, Boal DK, et al.: Fetal rhabdomyoma and nevoid basal cell carcinoma syndrome. Pediatr Pathol 12 (3): 441-7, 1992 May-Jun.
- Korczak JF, Brahim JS, DiGiovanna JJ, et al.: Nevoid basal cell carcinoma syndrome with medulloblastoma in an African-American boy: a rare case illustrating gene-environment interaction. Am J Med Genet 69 (3): 309-14, 1997.
- Wolthers OD, Stellfeld M: Benign mesenchymoma in the trachea of a patient with the nevoid basal cell carcinoma syndrome. J Laryngol Otol 101 (5): 522-6, 1987.
- Ponti G, Manfredini M, Pastorino L, et al.: PTCH1 Germline Mutations and the Basaloid Follicular Hamartoma Values in the Tumor Spectrum of Basal Cell Carcinoma Syndrome (NBCCS). Anticancer Res 38 (1): 471-476, 2018.
- Iacono RP, Apuzzo ML, Davis RL, et al.: Multiple meningiomas following radiation therapy for medulloblastoma. Case report. J Neurosurg 55 (2): 282-6, 1981.
- Mack EE, Wilson CB: Meningiomas induced by high-dose cranial irradiation. J Neurosurg 79 (1): 28-31, 1993.
- Moss SD, Rockswold GL, Chou SN, et al.: Radiation-induced meningiomas in pediatric patients. Neurosurgery 22 (4): 758-61, 1988.
- Chiritescu E, Maloney ME: Acrochordons as a presenting sign of nevoid basal cell carcinoma syndrome. J Am Acad Dermatol 44 (5): 789-94, 2001.
- Tom WL, Hurley MY, Oliver DS, et al.: Features of basal cell carcinomas in basal cell nevus syndrome. Am J Med Genet A 155A (9): 2098-104, 2011.
- Lo Muzio L, Nocini PF, Savoia A, et al.: Nevoid basal cell carcinoma syndrome. Clinical findings in 37 Italian affected individuals. Clin Genet 55 (1): 34-40, 1999.
- Goldstein AM, Pastakia B, DiGiovanna JJ, et al.: Clinical findings in two African-American families with the nevoid basal cell carcinoma syndrome (NBCC). Am J Med Genet 50 (3): 272-81, 1994.
- Yasar B, Byers HJ, Smith MJ, et al.: Common variants modify the age of onset for basal cell carcinomas in Gorlin syndrome. Eur J Hum Genet 23 (5): 708-10, 2015.
- Mazzola CA, Pollack IF: Medulloblastoma. Curr Treat Options Neurol 5 (3): 189-198, 2003.
- Amlashi SF, Riffaud L, Brassier G, et al.: Nevoid basal cell carcinoma syndrome: relation with desmoplastic medulloblastoma in infancy. A population-based study and review of the literature. Cancer 98 (3): 618-24, 2003.
- Cowan R, Hoban P, Kelsey A, et al.: The gene for the naevoid basal cell carcinoma syndrome acts as a tumour-suppressor gene in medulloblastoma. Br J Cancer 76 (2): 141-5, 1997.
- Evans DG, Farndon PA, Burnell LD, et al.: The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer 64 (5): 959-61, 1991.
- Berlin NI, Van Scott EJ, Clendenning WE, et al.: Basal cell nevus syndrome. Combined clinical staff conference at the National Institutes of Health. Ann Intern Med 64 (2): 403-21, 1966.
- Jackson R, Gardere S: Nevoid basal cell carcinoma syndrome. Can Med Assoc J 105 (8): 850 passim, 1971.
- Lindeberg H, Halaburt H, Larsen PO: The naevoid basal cell carcinoma syndrome. Clinical, biochemical and radiological aspects. J Maxillofac Surg 10 (4): 246-9, 1982.
- CAWSON RA, KERR GA: THE SYNDROME OF JAW CYSTS, BASAL CELL TUMOURS AND SKELETAL ABNORMALITIES. Proc R Soc Med 57: 799-801, 1964.
- Kedem A, Even-Paz Z, Freund M: Basal cell nevus syndrome associated with malignant melanoma of the iris. Dermatologica 140 (2): 99-106, 1970.
- Zvulunov A, Strother D, Zirbel G, et al.: Nevoid basal cell carcinoma syndrome. Report of a case with associated Hodgkin's disease. J Pediatr Hematol Oncol 17 (1): 66-70, 1995.
- Potaznik D, Steinherz P: Multiple nevoid basal cell carcinoma syndrome and Hodgkin's disease. Cancer 53 (12): 2713-5, 1984.
- Beddis IR, Mott MG, Bullimore J: Case report: nasopharyngeal rhabdomyosarcoma and Gorlin's naevoid basal cell carcinoma syndrome. Med Pediatr Oncol 11 (3): 178-9, 1983.
- Sobota A, Pena M, Santi M, et al.: Undifferentiated sinonasal carcinoma in a patient with nevoid basal cell carcinoma syndrome. Int J Surg Pathol 15 (3): 303-6, 2007.
- González-Alva P, Tanaka A, Oku Y, et al.: Keratocystic odontogenic tumor: a retrospective study of 183 cases. J Oral Sci 50 (2): 205-12, 2008.
- Suzuki M, Nagao K, Hatsuse H, et al.: Molecular pathogenesis of keratocystic odontogenic tumors developing in nevoid basal cell carcinoma syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol 116 (3): 348-53, 2013.
- Shear M: The aggressive nature of the odontogenic keratocyst: is it a benign cystic neoplasm? Part 1. Clinical and early experimental evidence of aggressive behaviour. Oral Oncol 38 (3): 219-26, 2002.
- Guo YY, Zhang JY, Li XF, et al.: PTCH1 gene mutations in Keratocystic odontogenic tumors: a study of 43 Chinese patients and a systematic review. PLoS One 8 (10): e77305, 2013.
- Gu XM, Zhao HS, Sun LS, et al.: PTCH mutations in sporadic and Gorlin-syndrome-related odontogenic keratocysts. J Dent Res 85 (9): 859-63, 2006.
- Lam KY, Chan AC: Odontogenic keratocysts: a clinicopathological study in Hong Kong Chinese. Laryngoscope 110 (8): 1328-32, 2000.
- North JP, McCalmont TH, LeBoit P: Palmar pits associated with the nevoid basal cell carcinoma syndrome. J Cutan Pathol 39 (8): 735-8, 2012.
- Chenevix-Trench G, Wicking C, Berkman J, et al.: Further localization of the gene for nevoid basal cell carcinoma syndrome (NBCCS) in 15 Australasian families: linkage and loss of heterozygosity. Am J Hum Genet 53 (3): 760-7, 1993.
- Ratcliffe JF, Shanley S, Ferguson J, et al.: The diagnostic implication of falcine calcification on plain skull radiographs of patients with basal cell naevus syndrome and the incidence of falcine calcification in their relatives and two control groups. Br J Radiol 68 (808): 361-8, 1995.
- Ratcliffe JF, Shanley S, Chenevix-Trench G: The prevalence of cervical and thoracic congenital skeletal abnormalities in basal cell naevus syndrome; a review of cervical and chest radiographs in 80 patients with BCNS. Br J Radiol 68 (810): 596-9, 1995.
- Betancourt NJ, Qian MF, Pickford JR, et al.: Gorlin Syndrome: Assessing Genotype-Phenotype Correlations and Analysis of Early Clinical Characteristics as Risk Factors for Disease Severity. J Clin Oncol 40 (19): 2119-2127, 2022.
- Fujii K, Ohashi H, Suzuki M, et al.: Frameshift mutation in the PTCH2 gene can cause nevoid basal cell carcinoma syndrome. Fam Cancer 12 (4): 611-4, 2013.
- Muller EA, Aradhya S, Atkin JF, et al.: Microdeletion 9q22.3 syndrome includes metopic craniosynostosis, hydrocephalus, macrosomia, and developmental delay. Am J Med Genet A 158A (2): 391-9, 2012.
- Huq AJ, Walsh M, Rajagopalan B, et al.: Mutations in SUFU and PTCH1 genes may cause different cutaneous cancer predisposition syndromes: similar, but not the same. Fam Cancer 17 (4): 601-606, 2018.
- Brugières L, Remenieras A, Pierron G, et al.: High frequency of germline SUFU mutations in children with desmoplastic/nodular medulloblastoma younger than 3 years of age. J Clin Oncol 30 (17): 2087-93, 2012.
- Aavikko M, Li SP, Saarinen S, et al.: Loss of SUFU function in familial multiple meningioma. Am J Hum Genet 91 (3): 520-6, 2012.
- DiGiovanna JJ, Kraemer KH: Shining a light on xeroderma pigmentosum. J Invest Dermatol 132 (3 Pt 2): 785-96, 2012.
- Michaëlsson G, Olsson E, Westermark P: The Rombo syndrome: a familial disorder with vermiculate atrophoderma, milia, hypotrichosis, trichoepitheliomas, basal cell carcinomas and peripheral vasodilation with cyanosis. Acta Derm Venereol 61 (6): 497-503, 1981.
- van Steensel MA, Jaspers NG, Steijlen PM: A case of Rombo syndrome. Br J Dermatol 144 (6): 1215-8, 2001.
- Ashinoff R, Jacobson M, Belsito DV: Rombo syndrome: a second case report and review. J Am Acad Dermatol 28 (6): 1011-4, 1993.
- Viksnins P, Berlin A: Follicular atrophoderma and basal cell carcinomas: the Bazex syndrome. Arch Dermatol 113 (7): 948-51, 1977.
- Vabres P, de Prost Y: Bazex-Dupré-Christol syndrome: a possible diagnosis for basal cell carcinomas, coarse sparse hair, and milia. Am J Med Genet 45 (6): 786, 1993.
- Rapelanoro R, Taïeb A, Lacombe D: Congenital hypotrichosis and milia: report of a large family suggesting X-linked dominant inheritance. Am J Med Genet 52 (4): 487-90, 1994.
- Vabres P, Lacombe D, Rabinowitz LG, et al.: The gene for Bazex-Dupré-Christol syndrome maps to chromosome Xq. J Invest Dermatol 105 (1): 87-91, 1995.
- Parren LJ, Abuzahra F, Wagenvoort T, et al.: Linkage refinement of Bazex-Dupré-Christol syndrome to an 11·4-Mb interval on chromosome Xq25-27.1. Br J Dermatol 165 (1): 201-3, 2011.
- Parrish JA, Baden HP, Goldsmith LA, et al.: Studies of the density and the properties of the hair in a new inherited syndrome of hypotrichosis. Ann Hum Genet 35 (3): 349-56, 1972.
- Gould DJ, Barker DJ: Follicular atrophoderma with multiple basal cell carcinomas (Bazex). Br J Dermatol 99 (4): 431-5, 1978.
- Yung A, Newton-Bishop JA: A case of Bazex-Dupré-Christol syndrome associated with multiple genital trichoepitheliomas. Br J Dermatol 153 (3): 682-4, 2005.
- Kidd A, Carson L, Gregory DW, et al.: A Scottish family with Bazex-Dupré-Christol syndrome: follicular atrophoderma, congenital hypotrichosis, and basal cell carcinoma. J Med Genet 33 (6): 493-7, 1996.
- Arin MJ, Grimberg G, Schumann H, et al.: Identification of novel and known KRT5 and KRT14 mutations in 53 patients with epidermolysis bullosa simplex: correlation between genotype and phenotype. Br J Dermatol 162 (6): 1365-9, 2010.
- Has C, Bauer JW, Bodemer C, et al.: Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol 183 (4): 614-627, 2020.
- Fine JD: Inherited epidermolysis bullosa. Orphanet J Rare Dis 5: 12, 2010.
- Fine JD: Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates From the National Epidermolysis Bullosa Registry. JAMA Dermatol 152 (11): 1231-1238, 2016.
- Fine JD, Johnson LB, Weiner M, et al.: Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986-2006. J Am Acad Dermatol 60 (2): 203-11, 2009.
- García M, Santiago JL, Terrón A, et al.: Two novel recessive mutations in KRT14 identified in a cohort of 21 Spanish families with epidermolysis bullosa simplex. Br J Dermatol 165 (3): 683-92, 2011.
- Bolling MC, Lemmink HH, Jansen GH, et al.: Mutations in KRT5 and KRT14 cause epidermolysis bullosa simplex in 75% of the patients. Br J Dermatol 164 (3): 637-44, 2011.
- Johnson RL, Rothman AL, Xie J, et al.: Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272 (5268): 1668-71, 1996.
- Hahn H, Wicking C, Zaphiropoulous PG, et al.: Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 85 (6): 841-51, 1996.
- Scheinfeld N, Hu G, Gill M, et al.: Identification of a recurrent mutation in the CYLD gene in Brooke-Spiegler syndrome. Clin Exp Dermatol 28 (5): 539-41, 2003.
- Bignell GR, Warren W, Seal S, et al.: Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet 25 (2): 160-5, 2000.
- Weyers W, Nilles M, Eckert F, et al.: Spiradenomas in Brooke-Spiegler syndrome. Am J Dermatopathol 15 (2): 156-61, 1993.
- Rajan N, Langtry JA, Ashworth A, et al.: Tumor mapping in 2 large multigenerational families with CYLD mutations: implications for disease management and tumor induction. Arch Dermatol 145 (11): 1277-84, 2009.
- Requena L, Fariña MC, Robledo M, et al.: Multiple hereditary infundibulocystic basal cell carcinomas: a genodermatosis different from nevoid basal cell carcinoma syndrome. Arch Dermatol 135 (10): 1227-35, 1999.
- Nordin H, Månsson T, Svensson A: Familial occurrence of eccrine tumours in a family with ectodermal dysplasia. Acta Derm Venereol 68 (6): 523-30, 1988.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Basal Cell Skin Cancer. Version 2.2022. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2022. Available online with free subscription Last accessed August 22, 2023.
- Foulkes WD, Kamihara J, Evans DGR, et al.: Cancer Surveillance in Gorlin Syndrome and Rhabdoid Tumor Predisposition Syndrome. Clin Cancer Res 23 (12): e62-e67, 2017.
- Lin JS, Eder M, Weinmann S, et al.: Behavioral Counseling to Prevent Skin Cancer: Systematic Evidence Review to Update the 2003 U.S. Preventive Services Task Force Recommendation. Agency for Healthcare Research and Quality, 2011. Report No.: 11-05152-EF-1. Also available online. Last accessed December 8, 2023.
- Peck GL, DiGiovanna JJ, Sarnoff DS, et al.: Treatment and prevention of basal cell carcinoma with oral isotretinoin. J Am Acad Dermatol 19 (1 Pt 2): 176-85, 1988.
- Goldberg LH, Hsu SH, Alcalay J: Effectiveness of isotretinoin in preventing the appearance of basal cell carcinomas in basal cell nevus syndrome. J Am Acad Dermatol 21 (1): 144-5, 1989.
- Cristofolini M, Zumiani G, Scappini P, et al.: Aromatic retinoid in the chemoprevention of the progression of nevoid basal-cell carcinoma syndrome. J Dermatol Surg Oncol 10 (10): 778-81, 1984.
- Tang JY, Mackay-Wiggan JM, Aszterbaum M, et al.: Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N Engl J Med 366 (23): 2180-8, 2012.
- Tang JY, Ally MS, Chanana AM, et al.: Inhibition of the hedgehog pathway in patients with basal-cell nevus syndrome: final results from the multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 17 (12): 1720-1731, 2016.
- Dréno B, Kunstfeld R, Hauschild A, et al.: Two intermittent vismodegib dosing regimens in patients with multiple basal-cell carcinomas (MIKIE): a randomised, regimen-controlled, double-blind, phase 2 trial. Lancet Oncol 18 (3): 404-412, 2017.
- Chen AC, Martin AJ, Choy B, et al.: A Phase 3 Randomized Trial of Nicotinamide for Skin-Cancer Chemoprevention. N Engl J Med 373 (17): 1618-26, 2015.
- Stockfleth E, Ulrich C, Hauschild A, et al.: Successful treatment of basal cell carcinomas in a nevoid basal cell carcinoma syndrome with topical 5% imiquimod. Eur J Dermatol 12 (6): 569-72, 2002 Nov-Dec.
- Mougel F, Debarbieux S, Ronger-Savlé S, et al.: Methylaminolaevulinate photodynamic therapy in patients with multiple basal cell carcinomas in the setting of Gorlin-Goltz syndrome or after radiotherapy. Dermatology 219 (2): 138-42, 2009.
- Basset-Seguin N, Bissonnette R, Girard C, et al.: Consensus recommendations for the treatment of basal cell carcinomas in Gorlin syndrome with topical methylaminolaevulinate-photodynamic therapy. J Eur Acad Dermatol Venereol 28 (5): 626-32, 2014.
- Skvara H, Kalthoff F, Meingassner JG, et al.: Topical treatment of Basal cell carcinomas in nevoid Basal cell carcinoma syndrome with a smoothened inhibitor. J Invest Dermatol 131 (8): 1735-44, 2011.
- Goldberg LH, Landau JM, Moody MN, et al.: Resolution of odontogenic keratocysts of the jaw in basal cell nevus syndrome with GDC-0449. Arch Dermatol 147 (7): 839-41, 2011.
- Ally MS, Tang JY, Joseph T, et al.: The use of vismodegib to shrink keratocystic odontogenic tumors in patients with basal cell nevus syndrome. JAMA Dermatol 150 (5): 542-5, 2014.
Squamous Cell Carcinoma
Introduction
Squamous cell carcinoma (SCC) is the second most common type of skin cancer and accounts for approximately 20% of cutaneous malignancies. Although most cancer registries do not include information on the incidence of keratinocyte carcinomas (basal cell carcinoma [BCC] and SCC), annual incidence estimates range from 1 million to 5.4 million cases in the United States. Multiple studies indicate an increased risk of SCC after a first keratinocyte carcinoma; a meta-analysis and review of 45 studies estimated that after a primary SCC diagnosis, 13.3% of individuals would develop a second SCC (95% confidence interval [CI], 7.4%–22.8%).
Mortality is rare from this cancer; however, the morbidity and costs associated with its treatment are considerable.
Risk Factors for Squamous Cell Carcinoma
Sun exposure and other risk factors
Sun exposure is the major known environmental factor associated with the development of skin cancer of all types; however, different patterns of sun exposure are associated with each major type of skin cancer. Unlike BCC, SCC is associated with chronic exposure, rather than intermittent, intense exposure to ultraviolet (UV) radiation. Occupational exposure is the characteristic pattern of sun exposure that is associated with SCC. Other agents and factors associated with SCC risk include tanning beds, arsenic, therapeutic radiation (such as psoralen and UVA therapy for psoriasis), chronic skin ulceration, and immunosuppression. For more information about exposures that can cause skin cancer in the general population, see Skin Cancer Prevention.
Characteristics of the skin
Like melanoma and BCC, SCC occurs more frequently in individuals with lighter skin than in those with darker skin. A case-control study of 415 cases and 415 controls showed similar findings; relative to Fitzpatrick type I skin, individuals with increasingly darker skin had decreased risks of skin cancer (odds ratios [ORs], 0.6, 0.3, and 0.1, for Fitzpatrick types II, III, and IV, respectively). The same study found that blue eyes and blond/red hair were also associated with increased risks of SCC, with crude ORs of 1.7 (95% CI, 1.2–2.3) for blue eyes, 1.5 (95% CI, 1.1–2.1) for blond hair, and 2.2 (95% CI, 1.5–3.3) for red hair. For more information, see the section on Pigmentary characteristics in the Melanoma section.
However, SCC can also occur in individuals with darker skin. An Asian registry based in Singapore reported an increase in skin cancer in that geographic area, with an incidence rate of 8.9 per 100,000 person-years. Incidence of SCC, however, was shown to be on the decline. SCC is the most common form of skin cancer in Black individuals in the United States and in certain parts of Africa; the mortality rate for this disease is relatively high in these populations. Epidemiologic characteristics of, and prevention strategies for, SCC in those individuals with darker skin remain areas of investigation.
Freckling of the skin and reaction of the skin to sun exposure have been identified as other risk factors for SCC. Individuals with heavy freckling on the forearm were found to have about a fourfold increase in SCC risk if moderate or heavy freckling was present in childhood or as an adult. The degree of SCC risk corresponded to the amount of freckling. In this study, the inability of the skin to tan and its propensity to burn were also significantly associated with risk of SCC (OR of 1.54 for burn/blisters and 12.44 for freckles/no tan).
The presence of scars on the skin can also increase the risk of SCC, although the process of carcinogenesis in this setting may take years or even decades. SCCs arising in chronic wounds are referred to as Marjolin’s ulcers. The mean time for development of carcinoma in these wounds is estimated at 26 years. One case report documents the occurrence of cancer in a wound that was incurred 59 years earlier.
Immunosuppression
Immunosuppression also contributes to the formation of BCCs and SCCs. Among solid-organ transplant recipients, the risk of SCC is 65 to 250 times higher, and the risk of BCC is 10 times higher than that observed in the general population, although the risks vary with transplant type and with the immunosuppressive agent used. BCCs and SCCs in high-risk patients (solid-organ transplant recipients and chronic lymphocytic leukemia patients) occur at a younger age, are more common and more aggressive, and have a higher risk of recurrence and metastatic spread than these cancers do in the general population. Additionally, there is a high risk of second SCCs. In one study, more than 65% of kidney transplant recipients developed subsequent SCCs after their first diagnosis. Among Medicare patients with an intact immune system, BCCs occur as frequently as SCCs; in transplant patients, SCCs outnumber BCCs by a 2:1 ratio.
Personal history of BCC, SCC, and melanoma skin cancers
A personal history of BCC or SCC is strongly associated with subsequent SCC. A study from Ireland showed that individuals with a history of BCC had a 14% higher incidence of subsequent SCC; for men with a history of BCC, the subsequent SCC risk was 27% higher. In the same report, individuals with melanoma were also 2.5 times more likely to report a subsequent SCC. There is an approximate 20% increased risk of a subsequent lesion within the first year after a skin cancer has been diagnosed. The mean age of occurrence for these cancers is the middle of the sixth decade of life.
A Swedish study of 224 melanoma probands and 944 of their first-degree relatives (FDRs) from 154 CDKN2A wild-type families and 11,680 matched controls showed that personal and family histories of melanoma increased the risk of SCC, with relative risks (RRs) of 9.1 (95% CI, 6.0–13.7) for personal history and 3.4 (95% CI, 2.2–5.2) for family history. Another study of 216,115 individuals from the Nurses’ Health Study, the Nurses’ Health Study 2, and the Health Professionals Follow-Up Study found that individuals with a family history of melanoma showed a 22% increased risk of SCC (hazard ratio,1.22; 95% CI, 1.06–1.40).
Family history of squamous cell carcinoma or associated premalignant lesions
Although the literature is scant on this subject, a family history of SCC may increase the risk of SCC in FDRs. In an independent survey-based study of 415 SCC cases and 415 controls, SCC risk was increased in individuals with a family history of SCC (adjusted OR, 3.4; 95% CI, 1.0–11.6), even after adjustment for skin type, hair color, and eye color. This risk was elevated to an OR of 5.6 in those with a family history of melanoma (95% CI, 1.6–19.7), 9.8 in those with a family history of BCC (95% CI, 2.6–36.8), and 10.5 in those with a family history of multiple types of skin cancer (95% CI, 2.7–29.6). Review of the Swedish Family Center Database showed that individuals with at least one sibling or parent affected with SCC, in situ SCC (Bowen disease), or actinic keratosis had a twofold to threefold increased risk of invasive and in situ SCC relative to the general population. Increased number of tumors in parents was associated with increased risk to the offspring. Of note, diagnosis of the proband at an earlier age was not consistently associated with a trend of increased incidence of SCC in the FDR, as would be expected in most hereditary syndromes because of germlinepathogenic variants. Further analysis of the Swedish population-based data estimates genetic risk effects of 8% and familial shared-environmental effects of 18%. Thus, shared environmental and behavioral factors likely account for some of the observed familial clustering of SCC.
A study on the heritability of cancer among 80,309 monozygotic and 123,382 dizygotic twins showed that NMSCs have a heritability of 43% (95% CI, 26%–59%), suggesting that almost half of the risk of NMSC is caused by inherited factors. Additionally, the cumulative risk of NMSC was 1.9-fold higher for monozygotic than for dizygotic twins (95% CI, 1.8–2.0).
Syndromes and Genes Associated With a Predisposition for Squamous Cell Carcinoma
Major genes have been defined elsewhere in this summary as genes that are necessary and sufficient for disease, with important pathogenic variants of the gene as causal. The disorders resulting from single-gene pathogenic variants within families lead to a very high risk of disease and are relatively rare. The influence of the environment on the development of disease in individuals with these single-gene disorders is often very difficult to determine because of the rarity of the genetic variant.
Identification of a strong environmental risk factor—chronic exposure to UV radiation—makes it difficult to apply genetic causation for SCC of the skin. Although the risk of UV exposure is well known, quantifying its attributable risk to cancer development has proven challenging. In addition, ascertainment of cases of SCC of the skin is not always straightforward. Many registries and other epidemiologic studies do not fully assess the incidence of SCC of the skin owing to: (1) the common practice of treating lesions suspicious for SCC without a diagnostic biopsy, and (2) the relatively low potential for metastasis. Moreover, NMSC is routinely excluded from the major cancer registries such as the Surveillance, Epidemiology, and End Results registry.
With these considerations in mind, the discussion below will address genes associated with disorders that have an increased incidence of skin cancer.
Characteristics of the major hereditary syndromes associated with a predisposition to SCC are described in Table 5 below.
| Condition | Gene(s) | Pathway | |
|---|---|---|---|
| SWI/SNF = SWItch/Sucrose Non-Fermentable. | |||
| aInformation from Loh et al. | |||
| Bloom syndrome | BLM/RECQL3 | Chromosomal stability | |
| Chediak-Higashi syndrome | LYST | Lysosomal transport regulation | |
| Dyskeratosis congenita | DKC1, TERC, TINF2, NHP2/NOLA2, NOP10/NOLA3, TERT, WRAP53, C16orf57, RTEL1 | Telomere maintenance and trafficking | |
| Dystrophic epidermolysis bullosa (autosomal dominant and autosomal recessive subtypes) | COL7A1 | Collagen anchor of basement membrane to dermis | |
| Elejalde disease | MYO5A | Pigment granule transport | |
| Epidermodysplasia verruciformis | EVER1/TMC6, EVER2/TMC8 | Signal transduction in endoplasmic reticulum | |
| Fanconi anemia | FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG/XRCC9, FANCI, FANCJ/BRIP1/BACH1, FANCL, FANCM, FANCN/PALB2, FANCO/RAD51C, FANCP/SLX4/BTBD12, FANCQ/ERCC4/XPF, FANCS/BRCA1 | DNA repair | |
| Griscelli syndrome (type 1, type 2, and type 3) | MYO5A, RAB27A, MLPH | Pigment granule transport | |
| Hermansky-Pudlak syndrome | HPS1, HPS2/AP3B1, HPS3, HPS4, HPS5, HPS6, HPS7/DTNBP1, HPS8/BLOC1S3, HPS9/BLOC1S6, HPS10/AP3D1 | Melanosomal and lysosomal storage | |
| Huriez syndromea | SMARCAD1 | SWI/SNF pathway chromatin regulator | |
| Junctional epidermolysis bullosa | LAMA3, LAMB3, LAMC2, COL17A1 | Connective tissue | |
| Multiple self-healing squamous epithelioma (Ferguson-Smith syndrome) | TGFBR1 | Growth factor signaling | |
| Oculocutaneous albinism (type IA, type IB, type II, type III, type IV, type V, type VI, and type VII) | TYR, OCA2, TYRP1, SLC45A2/MATP/OCA4, Locus 4q24, SLC24A5, C10Orf11 | Melanin synthesis | |
| Rothmund-Thomson syndrome type 1 | ANAPC1 | Cell cycle | |
| Rothmund-Thomson syndrome type 2 | RECQL4, C16orf57 | Chromosomal stability | |
| Werner syndrome | WRN/RECQL2 | Chromosomal stability | |
| Xeroderma pigmentosum (complementation group A, group B, group C, group D, group E, group F, and group G) | XPA, XPB/ERCC3, XPC, XPD/ERCC2, XPE/DDB2, XPF/ERCC4, XPG/ERCC5 | Nucleotide excision repair | |
| Xeroderma pigmentosum variant | POLH | Error-prone polymerase | |
Xeroderma pigmentosum
Xeroderma pigmentosum (XP) is a hereditary disorder of nucleotide excision repair that results in cutaneous malignancies in the first decade of life. Affected individuals have an increased sensitivity to sunlight, resulting in a markedly increased risk of SCCs, BCCs, and melanomas. One report found that keratinocyte carcinomas (BCC and SCC) were increased 150-fold in individuals with XP; for those younger than 20 years, the prevalence has been estimated to be 5,000 to 10,000 times what would be expected in the general population.
The natural history of this disease begins in the first year of life, when sun sensitivity becomes apparent, and xerosis (dry skin) and pigmentary changes may occur in the sun-exposed skin. About one-half of XP patients have a history of severe burning on minimal sun exposure. Other XP patients do not have this reaction but develop freckle-like pigmentation before age 2 years on sun-exposed sites. These manifestations progress to skin atrophy and formation of telangiectasias. Approximately one-half of people with this disorder will develop BCC or SCC, and approximately one-quarter of these individuals will develop melanoma. In the absence of sun avoidance, the median age of diagnosis for any skin cancer is 8 to 9 years. On average, BCCs and SCCs occur at a younger age than melanoma in the XP population.
Noncutaneous manifestations of XP can include ophthalmologic, neurological, and aging abnormalities. Cornea and eyelids abnormalities in XP can be linked to UV radiation exposure. Examples of ophthalmologic abnormalities can include the following: keratitis, corneal opacification, ectropion, entropion, hyperpigmentation of the eyelids, loss of eyelashes, and cancer (including conjunctival and corneal cancers). About 25% of the patients with XP examined at the National Institutes of Health (NIH) between 1971 and 2009 had progressive neurological degeneration. Features of neurological degeneration can include the following: microcephaly, progressive sensorineural hearing loss, diminished deep tendon reflexes, seizures, and cognitive impairment. Neurological degeneration, which is most commonly observed in individuals with complementation groups XPA and XPD, was associated with a shorter lifespan (median age of death was 29 years in individuals with neurological degeneration and 37 years in individuals without neurological degeneration). A subset of patients who have the skin and eye manifestations associated with XP also exhibit severe neurological manifestations, dwarfism, and delayed development. This severe form of XP is known as De Sanctis-Cacchione syndrome. Individuals with XP may also experience differences with aging. In a small study, 18 postmenopausal women with XP had a high frequency of premature aging, with a median age of menopause occurring at 29.5 years in the XP group (range, 18.0–49.5 y) and 52.9 years in the U.S. general population.
A variety of noncutaneous neoplasms–most notably SCC on the tip of the tongue, central nervous system cancers, hematologic cancers, thyroid cancers, gynecologic cancers, and lung cancers in smokers–have been reported in people with XP. The RR for internal cancers is estimated to be 34- to 50-fold higher in individuals with XP than in individuals in the general population. A report on 434 patients with XP in four international cohorts (United States, United Kingdom, France, and Brazil) found an internal cancers rate of 11.3% in individuals with XP, with the average age of diagnosis occurring 50 years earlier than that of individuals in the general U.S. population. Patients with pathogenic variants in the XPC gene (especially those from North Africa with a pathogenic delTG founder variant) had higher cancer risks than patients with pathogenic variants in other XP genes.
The inheritance for XP is autosomal recessive. Seven complementation groups have been associated with this disorder. About 40% of the XP cases seen at the NIH were XPC. ERCC2 (XPD) pathogenic variants were present in about 20%. Complementation group A, due to a pathogenic variant in XPA, accounts for approximately 10% of cases. Other variant genes in this disorder include ERCC3 (XPB), DDB2 (XPE), ERCC4 (XPF), and ERCC5 (XPG). An XPH group had been described but is now considered to be a subgroup of the XPD group. Heterozygotes for pathogenic variants in XP genes are generally asymptomatic. However, one study reported a threefold increase in BCC in Japanese individuals who were heterozygous for XPA pathogenic variants. Founder pathogenic variants in XPA (R228A) and XPC (V548A fs X572) have been identified in North African populations, and a founder pathogenic variant in XPC resulting in a splice alteration (IVS 12-1G>C) has been found in an East African (Mahori) population. It has been proposed that direct screening for these pathogenic variants would be appropriate in these populations. A founder pathogenic variant at the 3’ splice acceptor site of intron 3 in the XPA gene is present in approximately 1% of the population in Japan representing nearly 1 million people.
The function of the XP genes is to recognize and repair photoproducts from UV radiation. The main photoproducts are formed at adjacent pyrimidines and consist of cyclobutane dimers and pyrimidine-pyrimidone (6-4) photoproducts. The product of XPC is involved in the initial identification of DNA damage; it binds to the lesion to act as a marker for further repair. The DDB2 (XPE) protein is also part of this process and works with XPC. The XPA gene product maintains single-strand regions during repair and works with the TFIIH transcription factor complex. The TFIIH complex includes the gene products of both ERCC3 (XPB) and ERCC2 (XPD), which function as DNA helicases in the unwinding of the DNA. The ERCC4 (XPF) and ERCC5 (XPG) proteins act as DNA endonucleases to create single-strand nicks in the 5’ and 3’ sides of the damaged DNA with resulting excision of about 28 to 30 nucleotides, including the photoproduct. DNA polymerases replace the lesion with the correct sequence, and a DNA ligase completes the repair.
An XP variant that is associated with pathogenic variants in POLH (XPV) is responsible for approximately 10% of reported cases. This gene encodes for the error-prone bypass polymerase (polymerase eta) which, unlike other genes associated with XP, is not involved in nucleotide excision repair. People with polymerase eta pathogenic variants have the same cutaneous and ocular findings as other XP patients but do not have progressive neurologic degeneration. A founder pathogenic variant resulting in a deletion of exon 10 was seen in 16 of 16 individuals from ten Tunisian consanguineous families.
Work on genotype-phenotype correlations among the XP complementation groups continues; however, evidence suggests that the specific pathogenic variant may have more influence on the phenotype than the complementation group. The main distinguishing features appear to be the presence or absence of burning on minimal sun exposure, skin cancer, and progressive neurologic abnormalities. All complementation groups are characterized by the presence of cutaneous neoplasias, but skin cancers may be more common in XPC, XPE, and XPV groups.. There is additional clinical variation within each complementation group. Mild to severe neurologic impairment has been described in individuals with XPA pathogenic variants. Individuals with XPA pathogenic variants in the DNA binding region (amino acids 98–219) may have a more severe presentation that includes neurological findings. Individuals within the XPC complementation group have higher incidences of ocular damage. A very small number of people in the XPB, XPD, and XPG complementation groups have been identified as having xeroderma pigmentosum-Cockayne syndrome (XP-CS) complex. These individuals have characteristics of both disorders, including an increased predisposition to cutaneous neoplasms and developmental delay, visual and hearing impairment, and central and peripheral nervous system dysfunction. It should be noted that people with Cockayne syndrome without XP do not appear to have an increased cancer risk. Similarly, trichothiodystrophy (TTD) is another genetic disorder that can occur in combination with XP. Individuals affected solely with TTD do not appear to have an increased cancer incidence, but some affected with XP/TTD have an increased risk of cutaneous neoplasia. The complementation groups connected with XP/TTD (XPD and XPB) and XP-CS (XPB, XPD, and XPG) are associated with defects in both transcription-coupled nucleotide excision repair and global genomic nucleotide excision repair. In contrast, XP complementation groups C and E have defects only in global genomic nucleotide excision repair. In addition, individuals in the XPA, XPD and XPG groups may exhibit severe neurologic abnormalities without symptoms of Cockayne syndrome or TTD. Cerebro-oculo-facio-skeletal syndrome, which has been described with some ERCC2 (XPD) or XP-CS pathogenic variants, does not appear to confer an increased risk of skin cancer.
The diagnosis of XP is made on the basis of clinical findings and family history. Functional assays to assess DNA repair capabilities after exposure to radiation have been developed, but these tests are currently not clinically available in the United States. Clinical genetic testing using sequence analysis to identify pathogenic variants is available for multiple XP-associated genes; the list can be found at the NIH Genetic Testing Registry.
Multiple self-healing squamous epitheliomata (Ferguson-Smith syndrome)
Multiple self-healing squamous epitheliomata (MSSE), or Ferguson-Smith syndrome, first described in 1934, is characterized by invasive skin tumors that are histologically identical to sporadic cutaneous SCC, but they resolve spontaneously without intervention. Linkage analysis of affected families showed association with the long arm of chromosome 9, and haplotype analysis localized the gene to 9q22.3 between D9S197 and D9S1809. Transforming growth factor beta-receptor 1 (TGFBR1) was identified through next-generation sequencing as the gene responsible for MSSE. Loss-of-function pathogenic variants in TGFBR1 have been identified in 18 of 22 affected families. Gain-of-function variants in TGFBR1 are associated with unrelated Marfan-like syndromes, such as Loeys-Dietz syndrome, which have no described increase in skin cancer risk.
Somatic loss of heterozygosity in Ferguson-Smith–related SCC has been demonstrated at this genomic location, suggesting that TGFBR1 can act as a tumor suppressor gene. The long arm of chromosome 9 has also been a site of interest in sporadic SCC. Up to 65% of sporadic SCCs have been found to have loss of heterozygosity at 9q22.3 between D9S162 and D9S165.
Oculocutaneous albinism
Albinism is a major risk factor for skin cancer in individuals of African ancestry. One report describing a cohort of 350 albinos in Tanzania found 104 cutaneous cancers; of these, 100 were SCCs, three were BCCs, and one was melanoma. The median age for this population was 10 years. Similar proportions of skin cancer diagnoses were observed in a Nigerian population, with 62% of dermatological malignancies diagnosed as SCC, 16% as melanoma, and 8% as BCC. Of note, some melanomas found in individuals with albinism do contain melanin.
SCC occurring at extremely early ages is a hallmark of oculocutaneous albinism. In a cohort of nearly 1,000 Nigerian patients with albinism, all had malignant or premalignant cutaneous lesions by age 20 years.
Two types of oculocutaneous albinism are known to be associated with increased risk of SCC of the skin. Oculocutaneous albinism type 1, or tyrosinase-related albinism, is caused by pathogenic variants in the tyrosinase gene, TYR, located on the long arm of chromosome 11. This type of albinism accounts for about one-half of cases in individuals of European ancestry. The OCA2 gene, also known as the P gene, is altered in oculocutaneous albinism type 2, or tyrosinase-positive albinism. Both disorders are autosomal recessive, with frequent compound heterozygosity. A 2.7 kb intragenic deletion in the OCA2 gene is a common founder pathogenic variant in sub-Saharan Africa and accounts for most OCA2-related albinism cases in this region.
Tyrosinase acts as the critical enzyme in the synthesis of melanin in melanocytes. A variant in this gene in oculocutaneous albinism type 1 produces proteins with minimal to no activity, corresponding to the OCA1B and OCA1A phenotypes, respectively. Individuals with OCA1B have light skin, hair, and eye coloring at birth but develop some pigment during their lifetimes, while the coloring of those with OCA1A does not darken with age.
The gene product of OCA2 is a protein found in the membrane of melanosomes. Its function is unknown, but it may play a role in maintaining the structure or pH of this environment. Murine models with variants in this gene had significantly decreased melanin production compared with normal controls. In one international study of individuals with albinism, biallelic variants in OCA2 were found in 17% of participants.
Genetic variants in SLC45A2 (MATP associated with OCA4), SLC24A5 (associated with OCA6), and TYRP1 (tyrosinase-related protein 1 associated with OCA3) are associated with less common types of oculocutaneous albinism. Reported incidences for these genes in an international population of patients with albinism are 7% for SLC45A2, 1% for TYRP1, and less than 0.5% for SLC24A5. SLC45A2 is found in 24% of oculocutaneous albinism cases in Japan, making it the most common type of albinism among Japanese individuals with identifiable variants. A study of 22 individuals of Italian ancestry without pathogenic variants in TYR, OCA2, or TYRP1 found 5 individuals with biallelic variants in SLC45A2, 4 of whom met clinical criteria for a diagnosis of oculocutaneous albinism. Collectively, more than 600 unique ocular albinism–related genetic variants have been identified. OCA4 is thought to be similar to other types of albinism, but the increased risk of SCC of the skin in people with these variants has not been quantified. Skin cancer was not identified in a study of 30 individuals with OCA4. Of note, a meta-analysis demonstrated that the SLC45A2 p.Phe374Leu variant was protective for melanoma, with an OR of 0.41 (95% CI, 0.33–0.50; P = 3.50 x 10-17). However, at this time, it should be noted that clinical testing is not routinely performed for protective variants.
Additional genes associated with oculocutaneous albinism have been found in small numbers of patients. OCA5, located on chromosome 4q24, has been identified in a Pakistani family, whereas OCA6 appears to be caused by pathogenic variants in SLC24A5 on chromosome 15q21. Pathogenic variants in C10orf11 (LRMDA) cause OCA7, which has been found in patients from the Faroe Islands and Denmark. Small numbers of pathogenic variant carriers have been reported to date. One woman with OCA6 had actinic keratosis, but the incidence of skin cancers in these populations is unknown.
Although oculocutaneous albinism is inherited as an autosomal recessive disorder in most instances, one study has found that heterozygous variants in genes such as TYR, OCA2, TYRP1, and SLC45A2 are overrepresented in families with multiple cases of melanoma. Further investigation is warranted to determine if these genes may be moderate penetrance melanoma susceptibility genes in heterozygotes.
| Type | Subtype | Gene | Reporting Population | Availability of Clinical Test |
|---|---|---|---|---|
| OCA Type 1 | 1A | TYR | Japanese, Chinese, White | Yes |
| 1B | TYR | |||
| OCA Type 2 | OCA2 ( Pgene) | African, African American, American Indian | Yes | |
| OCA Type 3 | TYRP1 | African | Yes | |
| OCA Type 4 | SLC45A2 (MATP) | Japanese, Italian, German | Yes | |
| OCA Type 5 | OCA5 | Pakistani | Not in the United States | |
| OCA Type 6 | SLC24A5 | Chinese, African, European, Indian | Yes | |
| OCA Type 7 | C10orf11 (LRMDA) | Faroe Islands, Denmark | Yes |
Other albinism syndromes
A subgroup of albinism includes people who exhibit a triad of albinism, prolonged bleeding time, and deposition of a ceroid substance in organs such as the lungs and gastrointestinal tract. This syndrome, known as Hermansky-Pudlak syndrome, is inherited in an autosomal recessive manner but may have a pseudodominant inheritance in Puerto Rican families, owing to the high prevalence in this population. The underlying cause is believed to be a defect in melanosome and lysosome transport. A number of pathogenic variants at disparate loci have been associated with this syndrome, including HPS1, HPS3, HPS4, HPS5, HPS6, HPS7 (DTNBP1), HPS8 (BLOC1S3), and HPS9 (PLDN). Pigmentation characteristics can vary significantly in this disorder, particularly among those with HPS1 pathogenic variants, and patients report darkening of the skin and hair as they age. In a small cohort of individuals with HPS1 variants, 3 out of 40 developed cutaneous SCCs, and an additional 3 had BCCs. Hermansky-Pudlak syndrome type 2, which includes increased susceptibility to infection resulting from congenital neutropenia, has been attributed to defects in AP3B1.
Two additional syndromes are associated with decreased pigmentation of the skin and eyes. The autosomal recessive Chediak-Higashi syndrome is characterized by eosinophilic, peroxidase-positive inclusion bodies in early leukocyte precursors, hemophagocytosis, increased susceptibility to infection, and increased incidence of an accelerated phase lymphohistiocytosis. Pathogenic variants in the LYST gene underlie this syndrome, which is often fatal in the first decade of life.
Griscelli syndrome, also inherited in an autosomal recessive manner, was originally described as decreased cutaneous pigmentation with hypomelanosis and neurologic deficits, but its clinical presentation is quite variable. This combination of symptoms is now designated Griscelli syndrome type 1 or Elejalde disease. It has been attributed to pathogenic variants in the MYO5A gene, which affects melanosome transport. Individuals with Griscelli syndrome type 2 have decreased cutaneous pigmentation and immunodeficiency but lack neurological deficits. They also may have hemophagocytosis or lymphohistiocytosis that is often fatal, like that seen in Chediak-Higashi syndrome. Griscelli syndrome type 2 is caused by pathogenic variants in RAB27A, which is part of the same melanosome transport pathway as MYO5A. Griscelli syndrome type 3 presents with hypomelanosis and does not include neurologic or immunologic disorders. Pathogenic variants in the melanophilin (MLPH) gene and MYO5A have been associated with this variant of Griscelli syndrome.
Epidermolysis bullosa
There are numerous forms of epidermolysis bullosa (EB), which is characterized by cleavage and blistering of the skin. A study from the Dutch EB registry found an overall EB incidence of 41.3 per million live births and 22.4 per million population. Similarly, a German study found an EB incidence of 45.1 per million live births. However, a study from England and Wales found a much higher EB incidence of 67.8 per million population. In the most extreme cases, EB can result in the congenital absence of the skin. Dystrophic EB and junctional EB are associated with an increased risk of skin cancer, particularly SCC. While the type of EB can be difficult to determine clinically, next-generation sequencing can define the EB subtype in up to 90% of cases. Diagnosis of EB may be accomplished by immunofluorescence or electron microscopy. The types of EB, pathogenic variants involved, and phenotypic characteristics are detailed in the following review. For more information, see the EB simplex section.
Dystrophic epidermolysis bullosa
Approximately 95% of individuals with the heritable disorder, dystrophic epidermolysis bullosa (DEB), have a detectable germline pathogenic variant in the COL7A1 gene. This gene (located at 3p21.3) is expressed in basal keratinocytes of the epidermis and encodes type VII collagen. This collagen forms a part of the fibrils that anchor the basement membrane to the dermis, thereby providing structural stability and resistance to mild skin trauma. A lack of type VII collagen often results in generalized skin blistering (starting at birth), skin atrophy, and scarring. A registry of DEB pathogenic variants, The International DEB Patient Registry, is accessible on the internet. An observational study analyzed individuals in the Dutch EB registry over a 30-year period. Results found that DEB comprises 34.7% of all EB diagnoses and has a point-prevalence of 8.3 per million population. Studies from the Netherlands, Germany, and England/Wales showed a range of DEB incidence from 14.1 to 26.1 per million live births.
There are two recessively inherited subtypes of DEB: severe generalized (RDEB-sev gen; previously named Hallopeau-Siemens type) and generalized other or generalized intermediate (RDEB-O; previously named non–Hallopeau-Siemens type); and a dominantly inherited form, dominant dystrophic epidermolysis bullosa (DDEB). These syndromes are rare. The prevalence per million individuals in the United States and incidence per million live births are 0.36 and 0.57 for RDEB-sev gen, 0.14 and 0.30 for RDEB-O, and 1.49 and 2.12 for DDEB, respectively. The clinical manifestations demonstrate a continuum of severity that complicates definitive diagnosis, especially early in life. The severe generalized subtype, associated with formation of pseudosyndactyly (a mitten-like deformity secondary to fusion of interdigital webbing) in early childhood, carries an SCC risk of up to 85% by age 45 years. These cancers arise in nonhealing wounds and usually metastasize to cause death within 5 years of the diagnosis of SCC. In one case series, SCC was the leading cause of death for the 15 patients with the severe generalized subtype. The incidence of SCC appears to be highest in the RDEB subtype. In a review of 69 articles that included all types of EB, 117 individuals with SCC were identified; 81 of these cases (69.2%) were in individuals with RDEB. In this group, the median age of diagnosis was 36 years (range, 6–71 y). A cohort study found that 18 of 283 (6.4%) individuals with RDEB had a diagnosis of SCC with a median age of diagnosis of 22.6 years (interquartile range, 20–27.7 y). Early mortality also has been observed in this disorder, with a mortality rate of up to 40% by age 30 years. An epidemiological study in Germany found that the mean age of death was 28.03 years in patients with DEB. Extracutaneous manifestations of RDEB-sev gen include short stature, anemia, strictures of the gastrointestinal and genitourinary tracts, and corneal scarring that may result in blindness.
The rate of de novo pathogenic variants for DDEB is approximately 30%; maternal germline mosaicism has also been reported. Glycine substitutions in exons 73 to 75 are the most common pathogenic variants in DDEB. G2034R and G2043R account for half of these variants. Less frequently, splice junction pathogenic variants and substitutions of glycine and other amino acids may cause the dominant form of DEB. In contrast, more than 400 pathogenic variants have been described for the two types of recessive EB. The recessive form of the disease is caused primarily by null variants, although amino acid substitutions, splice junction variants, and missense variants have also been reported. In-frame exon skipping may generate a partially functional protein in recessive disease. A founder pathogenic variant, c.6527insC (p.R525X), has been observed in 27 of 49 Spanish individuals with recessive DEB. A founder pathogenic variant in COL7A1, pVal769LeuFsXI, was identified in 11 of 15 families in Sfax, Southern Tunisia. Three of 12 individuals carrying at least one copy of this variant developed SCC, including two young-onset cases at ages 16 and 29 years. Genotype-phenotype correlations suggest an inverse correlation between the amount of functional protein and severity.
Pathogenic variants in COL7A1 result in abnormal triple helical coiling and decreased function, which causes increased skin fragility and blistering. In studies of Ras-driven carcinogenesis in RDEB-severe generalized keratinocytes, retention of the amino-terminal NC1, the first noncollagenous fragment of type VII collagen, is tumorigenic in mice. This retained sequence may mediate tumor-stroma interactions that promote carcinogenesis.
Junctional epidermolysis bullosa
Junctional epidermolysis bullosa (JEB) is an autosomal recessive type of EB with an estimated incidence of 2.68 per million live births and an estimated prevalence of 0.49 per million individuals in the United States. A Dutch EB registry study reported higher JEB prevalence estimates, with JEB occurring at a rate of 9.3 per million live births and 2.1 per million individuals in the Netherlands. In this study, approximately 19% of all EB diagnoses were JEB. Incidence of JEB in England and Wales (8.9 per million live births) was similar to those from the Netherlands and the U.S., but a study from Germany reported a higher incidence of JEB (14.23 per million live births). JEB results in considerable mortality, with approximately 50% of cases dying within the first year of life. Pathogenic variants in COL17A1 or in any of the genes encoding laminin 332 and its three subunits, previously known as laminin 5 (LAMA3, LAMB3, LAMC2) can result in JEB. Individuals with a severe form of JEB, called the Herlitz type, have an 18% cumulative risk of developing SCC by age 25 years. A study of COL17A1 in individuals with a milder subtype of JEB, called JEB-other, identified 85 pathogenic variants in 86 alleles from 43 individuals. In this study, total loss of COL17A1 protein staining was associated with a severe JEB phenotype.
Epidermodysplasia verruciformis
Pathogenic variants in either of two adjacent genes on chromosome 17q25 can cause epidermodysplasia verruciformis, a rare heritable disorder associated with increased susceptibility to human papillomavirus (HPV). Infection with certain HPV subtypes can lead to development of generalized nonresolving verrucous lesions, which develop into in situ and invasive SCCs in 30% to 60% of patients. Malignant transformation is thought to occur in about half of these lesions. Approximately 90% of these lesions are attributed to HPV types 5 and 8, although types 14, 17, 20, and 47 have occasionally been implicated. The association between HPV infection and increased risk of SCC has also been demonstrated in people without epidermodysplasia verruciformis; one case-control study found that HPV antibodies were found more frequently in the plasma of individuals with SCC (OR, 1.6; 95% CI, 1.2–2.3) than in plasma from cancer-free individuals.
The genes associated with this disorder, EVER1 and EVER2, were identified in 2002. The inheritance pattern of these genes appears to be autosomal recessive; however, autosomal dominant inheritance has also been reported. Both of these gene products are transmembrane proteins localized to the endoplasmic reticulum, and they likely function in signal transduction. This effect may be through regulation of zinc balance; it has been shown that these proteins form a complex with the zinc transporter 1 (ZnT-1), which is, in turn, blocked by certain HPV proteins.
A recent case-control study examined the effect of a specific EVER2polymorphism (rs7208422) on the risk of cutaneous SCC in 239 individuals with prior SCC and 432 controls. This polymorphism is a (A > T) coding single nucleotide variant in exon 8, codon 306 of the EVER2 gene. The frequency of the T allele among controls was 0.45. Homozygosity for the polymorphism caused a modest increase in SCC risk, with an adjusted OR of 1.7 (95% CI, 1.1–2.7) relative to wild-type homozygotes. In this study, those with one or more of the T alleles were also found to have increased seropositivity for any HPV and for HPV types 5 and 8, as compared with the wild type.
Some evidence suggests nonallelic heterogeneity in epidermodysplasia verruciformis. An individual born to consanguineous parents with epidermodysplasia verruciformis and additional bacterial and fungal infections was found to have homozygous R115X pathogenic variants in the MST1 gene. Another susceptibility locus associated with this disorder has been identified at chromosome regions 2p21-p24 through linkage analysis of an affected consanguineous family. Unlike those with pathogenic variants in the EVER1 and EVER2 genes, affected individuals linked to this genomic region were infected with HPV 20 rather than the usual HPV subtypes associated with this disorder, and this family did not have a history of cutaneous SCC.
Fanconi anemia
Fanconi anemia is a complex disorder that is characterized by increased incidence of hematologic and solid tumors, including SCC of the skin. Fanconi anemia is inherited as an autosomal recessive disease. It is a relatively rare syndrome with an estimated carrier frequency of one in 181 individuals in the United States (range: 1 in 156 to 1 in 209) and a carrier frequency of up to 1 in 100 individuals of Ashkenazi Jewish ancestry. Leukemia is the most commonly reported cancer in this population, but increased rates of gastrointestinal, head and neck, and gynecologic cancers have also been seen. By age 40 years, individuals affected with Fanconi anemia have an 8% risk per year of developing a solid tumor; the median age of diagnosis for solid tumors is 26 years. Multiple cases of cancers of the brain, breast, lung, and kidney (Wilms tumor) have been reported in this population. Data on the incidence of NMSCs in this population are sparse; however, review of the literature suggests that the age of diagnosis is between the mid-20s and early 30s and that women seem to be affected more often than men.
Individuals with this disease have increased susceptibility to DNA cross-linking agents (e.g., mitomycin-C or diepoxybutane) and ionizing and UV radiation. The diagnosis of this disease is made by observing increased chromosomal breakage, rearrangements, or exchanges in cells after exposure to carcinogens such as diepoxybutane.
Seventeen complementation groups have been identified for Fanconi anemia; details regarding the genes associated with these groups are listed in Table 7 below. Exome sequencing has revealed that a subset of individuals can carry multiple heterozygous pathogenic variants in Fanconi anemia genes, which may impact phenotypic presentation.
| Gene | Locus | Approximate Incidence Among FA Patients (%) | Pattern of Disease Transmission |
|---|---|---|---|
| AR = autosomal recessive; XLR = X-linked recessive. | |||
| FANCA | 16q24.3 | 70 | AR |
| FANCB | Xp22.31 | Rare | XLR |
| FANCC | 9q22.3 | 10 | AR |
| FANCD1 (BRCA2) | 13q12.3 | Rare | AR |
| FANCD2 | 3p25.3 | Rare | AR |
| FANCE | 6p21.3 | 10 | AR |
| FANCF | 11p15 | Rare | AR |
| FANCG (XRCC9) | 9p13 | 10 | AR |
| FANCI (KIAA1794) | 15q25-26 | Rare | AR |
| FANCJ (BACH1/BRIP1) | 17q22.3 | Rare | AR |
| FANCL (PHF9/POG) | 2p16.1 | Rare | AR |
| FANCM (Hef) | 14q21.3 | Rare | AR |
| FANCN (PALB2) | 16p12.1 | Rare | AR |
| FANCO (RAD51C) | 17q22 | Rare | AR |
| FANCP (SLX4/BTBD12) | 16p13.3 | Rare | AR |
| FANCQ (ERCC4/XPF) | 16p13.12 | Rare | AR |
| FANCS (BRCA1) | 17q21.31 | Rare | AR |
The proteins involved with DNA crosslink repairs are called the FANC pathway because of their involvement in Fanconi anemia. They interact with several other proteins associated with hereditary cancer risk, including those in Bloom syndrome and ataxia-telangiectasia. Further investigation has revealed that FANCD1 is the same gene as BRCA2, a gene that causes predisposition to breast and ovarian cancer. Other Fanconi anemia genes, FANCJ (BRIP1) and FANCN (PALB2), have also been identified as rare breast cancer susceptibility genes. For more information, see the Cancer Risks, Spectrum, and Characteristics section in BRCA1 and BRCA2: Cancer Risks and Management, or see the BRIP1, PALB2, and RAD51 sections in Genetics of Breast and Gynecologic Cancers. Individuals who are heterozygous carriers of other Fanconi anemia–associated variants do not appear to have an increased cancer risk, with the possible exception of a twofold increase in breast cancer incidence in carriers of FANCC pathogenic variants.
In 2018, a group reported a significant increase in SCC cases (OR, 1.69; 95% CI, 1.26–2.26) associated with a specific BRCA2 allele, which is relatively prevalent in the Icelandic population (K3326X; allele frequency, 1.1%). This allele results in normal production of an altered protein, and the authors hypothesized carriers have an increased sensitivity to environmental factors, which require DNA repair. This variant was also associated with an increased risk of small cell lung cancer, breast cancer, and ovarian cancer (but lower than the risk associated with the BRCA pathogenic variants that decrease protein levels).
Dyskeratosis congenita (Zinsser-Cole-Engman syndrome)
Dyskeratosis congenita, like Werner syndrome, results in premature aging and is thus considered a progeroid disease. The classic clinical triad for diagnosis includes nail dystrophy, reticular pigmentation of the chest and neck, and oral leukoplakia. In addition, individuals with this disorder are at markedly increased risk of myelodysplastic syndrome, acute leukemia, and bone marrow failure. Ocular, dental, neurologic, gastrointestinal, pulmonary, and skeletal abnormalities have also been described in conjunction with this disease, but clinical expressivity is variable. Developmental delay may also be present in variants of dyskeratosis congenita, such as Hoyeraal-Hreidarsson syndrome (HHS) and Revesz syndrome.
Approximately 10% of individuals with dyskeratosis congenita will develop nonhematologic tumors, often before the third decade of life. Solid tumors may be the first manifestation of this disorder. Head and neck cancers were the most commonly reported, accounting for nearly half of the cancers observed. Cutaneous SCC occurred in about 1.5% of the subjects, and the median age at diagnosis was 21 years. These cancers are generally managed as any other SCC of the skin.
Several genes associated with telomere function (DKC1, TERC, TINF2, NHP2, NOP10, RTEL1 and TERT) have been implicated in dyskeratosis congenita; approximately one-half of the individuals with a clinical diagnosis of this disease have an identified pathogenic variant in one of these seven genes. TERC and TINF2 are inherited in an autosomal dominant manner, whereas NHP2 (NOLA2) and NOP10 (NOLA3) show autosomal recessive inheritance, and RTEL1 and TERT can be either autosomal dominant or autosomal recessive. Recessive pathogenic variants in RTEL1 can also be associated with HHS. A study of more than 1,000 individuals of Ashkenazi Jewish ancestry identified a founder RTEL1 splice-site pathogenic variant, c.3791G>A (p.R1264H), that had a carrier frequency of 1% in Orthodox Ashkenazi Jewish individuals and 0.45% in the general Ashkenazi Jewish population. DKC1 shows an X-linked recessive pattern. Alterations in these genes result in shortening of telomeres, which in turn leads to defects in proliferation and spontaneous chromosomal rearrangements. Levels of TERC, the RNA component of the telomerase complex, are reduced in all dyskeratosis congenita patients. Missense pathogenic variants in WRAP53, a gene with a protein product that facilitates trafficking of telomerase, have also been associated with an autosomal recessive form of dyskeratosis congenita. Pathogenic variants in C16orf57 were identified in 6 of 132 families who did not have a variant detected in other known genes. C16orf57 pathogenic variants are also associated with poikiloderma with neutropenia. For more information about poikiloderma congenitale, see the Rothmund-Thomson syndrome section.
The recommended approach for diagnosis begins with a six-cell panel assay for leukocyte telomere length testing. If telomere length is in the lowest 1% for three or more cell types, molecular genetic testing is indicated. Testing of DKC1 may be performed first in male probands, as pathogenic variants in this gene account for up to 36% of those identified in dyskeratosis congenita to date. Pathogenic variants in TINF2 and TERT are responsible for 11% to 24% and 6% to 10% of cases, respectively.
Rothmund-Thomson syndrome
Rothmund-Thomson syndrome, also known as poikiloderma congenitale, is a heritable disorder characterized by chromosomal instability. The cutaneous presentation of this condition is an erythematous, blistering rash appearing on the face, buttocks, and extremities in early infancy. Other characteristics of this syndrome include telangiectasias, skeletal abnormalities, short stature, cataracts, and increased risk of osteosarcoma. Areas of hyperpigmentation and hypopigmentation of the skin develop later in life, and BCC or SCC can develop at an early age. Reports of multiple SCCs in situ have been reported in individuals as young as 16 years. The precise increased risk of skin cancer is not well characterized, but the point prevalence of keratinocyte carcinomas, including both BCC and SCC, is 2% to 5% in young individuals affected by this syndrome. This prevalence is clearly greater than that found in individuals in the same age group in the general population. Although increased UV sensitivity has been described, SCCs are also found in areas of the skin that are not exposed to the sun.
A pathogenic variant in the gene RECQL4 is present in 66% of clinically affected individuals with Rothmund-Thomson syndrome type 2. This gene is located at 8q24.3, and inheritance is believed to be autosomal recessive. RECQL4 encodes the ATP-dependent DNA helicase Q4, which promotes DNA unwinding to allow for cellular processes such as replication, transcription, and repair. A role for this protein in repair of DNA double-strand breaks has also been suggested. Pathogenic variants in similar DNA helicases lead to the inherited disorders of Bloom syndrome and Werner syndrome.
At least 19 different truncating pathogenic variants in this gene have been identified as deleterious. These pathogenic variants cause severe down-regulation of RECQL4 transcripts in this subset of individuals with Rothmund-Thomson syndrome. Cells deficient in RECQL4 have been found to be hypersensitive to oxidative stress, resulting in decreased DNA synthesis. Deficiencies in the RecQ helicases permit hyper-recombination, thereby leading to loss of heterozygosity. Loss of heterozygosity associated with deficiencies of this protein suggests that the helicases are caretaker-type tumor suppressor proteins.
Three of six families with Rothmund-Thomson syndrome type 2 were found to have homozygous pathogenic variants in the C16orf57 gene. Pathogenic variants in this gene have also been identified in individuals with dyskeratosis congenita and poikiloderma with neutropenia, suggesting that these syndromes are related; however, skin cancer risk in these conditions is not well characterized. For more information, see the Dyskeratosis congenita (Zinsser-Cole-Engman syndrome) section.
The clinical presentation of Rothmund-Thomson syndrome type 1 is similar to that of type 2, except that individuals present with bilateral juvenile cataracts, and they do not develop osteosarcoma. A study of seven families with ten children who have features of Rothmund-Thomson syndrome type 1 identified a deep intronic splicing variant in the ANAPC1 gene. Five individuals were homozygous for this variant, and three individuals had a second predicted pathogenic variant in ANAPC1 in trans. The splicing variant led to the introduction of a new exon, which caused premature termination codons with subsequent nonsense-mediated decay of ANAPC1 transcripts and reduced protein levels. ANAPC1 is a member of the anaphase-promoting complex/cyclosome.
Bloom syndrome
Loss of genomic stability is a major cause of Bloom syndrome. This disorder shows increased chromosomal breakage and is diagnosed by increased sister chromatid exchanges on chromosomal analysis. Clinical manifestations of Bloom syndrome include severe growth retardation, recurrent infections, diabetes, chronic pulmonary disease, and an increased susceptibility to cancers of many types. A study of 290 individuals from the Bloom Syndrome Registry (enrolled from 1960 to 2021) found that 155 participants (53%) developed one or more malignancies, with an 83% cumulative incidence of any cancer type by age 40 years. Median survival was 36.2 years. Median survival varied for those diagnosed with a hematologic malignancy (25.4 y) and those diagnosed with a solid tumor (37.7 y). Median survival was not adjusted by year of cancer diagnosis.
Skin cancer accounts for approximately 9% of tumors in the Bloom Syndrome Registry. Skin cancers occur at an early age in this population, with individuals diagnosed at a mean age of 31 years to 33 years. Almost 12% of registry participants developed a skin cancer, with BCC occurring most often. The mean age of skin cancer diagnosis was 33 years. The typical skin lesion seen in this disorder is a photosensitive erythematous telangiectatic rash that occurs in the first or second year of life. Although this rash is most commonly found on the face, it can also be present on the dorsa of the hands or forearms. SCC of the skin is the third most common malignancy associated with this disorder.
The BLM gene, located on the short arm of chromosome 15, is the only gene known to be associated with Bloom syndrome. This gene encodes a 1,417-amino acid protein that is regulated by the cell cycle and demonstrates DNA-dependent ATPase and DNA duplex-unwinding activities. Its helicase domain shows considerable similarity to the RecQ subfamily of DNA helicases. Absence of this gene product is thought to destabilize other enzymes that participate in DNA replication and repair.
This rare chromosomal breakage syndrome is inherited in an autosomal recessive manner and is characterized by loss of genomic stability. Sixty-four pathogenic variants described in the BLM gene include nucleotide insertions and deletions (41%), nonsense variants (30%), variants resulting in mis-splicing (14%), and missense variants (16%). A specific pathogenic variant identified in the Ashkenazi Jewish population is a 6-bp deletion/7-bp insertion at nucleotide 2,281, designated as BLMASH. Many of these variants result in truncation of the C-terminus, which prevents normal localization of this protein to the nucleus. Absence of functional BLM protein can cause increased rates of pathogenic variants and recombination. This somatic hypermutability leads to an increased risk of cancer at an early age in virtually every organ, including the skin.
Cells from people with Bloom syndrome have been found to have abnormal responses to UV radiation. Normal nuclear accumulation of TP53 after UV radiation was absent in 2 of 11 primary cultures from individuals with Bloom syndrome; in contrast, responses in cultures from people who have XP and ataxia-telangiectasia were normal. The gene product of the BLM gene has also been found to complex with Fanconi proteins, raising the possibility of connections between the BLM and Fanconi anemia pathways for DNA stability.
Werner syndrome
Like Bloom syndrome, Werner syndrome is characterized by spontaneous chromosomal instability, resulting in increased susceptibility to cancer and premature aging. Diagnostic criteria, often in the setting of consanguinity, include cataracts, short stature, premature graying or thinning of hair, and a positive 24-hour urinary hyaluronic acid test. Cardinal cutaneous manifestations of this disorder consist of sclerodermatous skin changes, ulcerations, atrophy, and pigmentation changes. Individuals with this syndrome have an average life expectancy of fewer than 50 years. Cancers have an early onset and occur in up to 43% of these patients. The spectrum of tumors associated with this disorder has primarily been described in the Japanese population and includes an increased incidence of sarcoma, thyroid cancers, and skin cancers. Approximately 20% of the cancers reported in this syndrome are cutaneous, with melanoma and SCC of the skin accounting for 14% and 5%, respectively. A study of 189 individuals with Werner syndrome estimated melanoma risk to be elevated 53-fold in these individuals. SCC was less frequently diagnosed. Acral lentiginous melanomas are overrepresented, and SCCs may exhibit more aggressive behavior, with metastasis to lymph nodes and internal organs.
Pathogenic variants in the WRN gene on chromosome 8p12-p11.2 have been identified in approximately 90% of individuals with this syndrome; no other genes are known to be associated with Werner syndrome. Inheritance of this gene is believed to be autosomal recessive. The product of the WRN gene is a multifunctional protein including a DNA exonuclease and an ATP-dependent DNA helicase belonging to the RecQ subfamily. This protein may play a role in processes such as DNA repair, recombination, replication, transcription, and combined DNA functions. Telomere dysfunction has been associated with premature aging and cancer susceptibility. Other helicases with similar function are altered in other chromosomal instability syndromes, such as BLM in Bloom syndrome and RecQL4 in Rothmund-Thomson syndrome.
Pathogenic variants described in the WRN gene include all types of variants, including missense, splice-site, and large rearrangements. A few founder pathogenic variants have been reported, which include a splice-site variant (previously known as IVS 25-1G→C; now known as c.3139-1G>C) found in over 50% of affected Japanese individuals.
Pathogenic variants in the WRN gene causes loss of nuclear localization of the gene product. Intracellular levels of the mRNA and protein associated with the variant are also markedly decreased, compared with those of the wild type. Half-lives of the mRNA and protein associated with the variant are also shorter than those associated with the wild-type mRNA and protein.
MC1R
A meta-analysis showed that the more MC1R pathogenic variants an individual carried, the higher his/her risk was to develop SCC and BCC. Individuals with two or more MC1R pathogenic variants had a summary OR of 2.48 (95% CI, 1.96–3.15) for BCC and a summary OR of 2.80 (95% CI, 1.71–4.57) for SCC; these risks increased when individuals had red hair. A study of individuals diagnosed with BCC before age 40 years also found a stronger association between BCC and MC1R pathogenic variants in those with phenotypic characteristics that are not traditionally considered high risk. For more information, see the section on MC1R in the Melanoma section.
Interventions
Prevention and treatment of skin cancers
A phase III, double-blind, placebo-controlled clinical trial in Australia evaluated the effects of oral nicotinamide (vitamin B3) in 386 individuals with a history of at least two NMSCs within 5 years before study enrollment. The mean age of the study participants was 66.4 years, and 63% were male. After 12 months of treatment, those taking nicotinamide 500 mg twice daily had a 30% reduction in the incidence of new SCCs (95% CI, 0%–51%; P = .05). A statistically significant reduction was also seen in actinic keratoses, the precursor skin lesions to SCCs. The rate of new NMSCs was 23% lower in the nicotinamide group (95% CI, 4%–38%, P = .02) than in the placebo group. No clinically significant differences in adverse events were observed between the two groups, and there was no evidence of benefit after discontinuation of nicotinamide. Of note, this study was not conducted in a population with an identified genetic predisposition to SCC, but it was conducted in a part of the world with a high incidence of sunlight-induced skin cancer.
Level of evidence (nicotinamide): 1aii
Because many of the syndromes described above are rare, few clinical trials have been conducted in these specific populations. However, valuable information has been developed from the clinical management experience related to skin cancer risk and treatment in the XP population. Strict sun avoidance beginning in infancy, use of protective clothing, and close clinical monitoring of the skin are key components to management of XP. Full-body photography of the skin, conjunctivae, and eyelids is recommended to aid in follow-up. Although few studies on treatment of SCC in the XP population have been done, in most cases treatment is similar to what would be recommended for the general population. Actinic keratoses are treated with topical therapies such as fluorouracil (5-FU), cryotherapy with liquid nitrogen, or dermabrasion, whereas cutaneous cancers are generally managed surgically.
Level of evidence: 5
Oral isotretinoin has been used as chemoprevention in XP patients with promising results. A small study of daily use of isotretinoin (13-cis retinoic acid; given as 2 mg/kg/day) reduced NMSC incidence by 63% in a small number of people with XP. Toxicities associated with this treatment included mucocutaneous symptoms, abnormalities in liver function tests and triglyceride levels, and musculoskeletal symptoms such as arthralgias, calcifications of tendons and ligaments, and osteoporosis. Dose reduction to 0.5 mg/kg/day reduced toxicity and decreased skin cancer frequency in three of seven subjects (43%); increasing the dose to 1 mg/kg/day resulted in decreased skin cancer frequency in three of the four subjects who did not respond at the lower dose. Oral isotretinoin use may be useful as a chemopreventive agent in other hereditary skin cancer syndromes, including basal cell nevus syndrome (BCNS), Rombo syndrome, EB, and epidermodysplasia verruciformis.
Level of evidence (oral isotretinoin for XP): 3aii
Level of evidence (oral isotretinoin for BCNS, Rombo syndrome, epidermodysplasia verruciformis): 5
Topical T4N5 liposome lotion, containing the bacterial enzyme T4 endonuclease V, was also investigated as a chemopreventive agent in a randomized, placebo-controlled trial of 30 XP patients. Although no effect was seen on incidence of SCC, 17.7 fewer actinic keratoses per year were seen in the treatment group. Additionally, 1.6 fewer BCCs per year were observed in patients being treated with this therapy. Both of these results were statistically significant. The risk of BCC was reduced by 47%, which was of borderline statistical significance. No significant adverse effects of this agent were reported. To date, this agent has not been approved for use by the U.S. Food and Drug Administration.
Level of evidence: 1aii
For patients with XP and unresectable SCC, therapy with 5-FU has been investigated. Several treatment methods were used in this prospective study, including topical therapy to the lesions, short systemic infusion with folic acid, and continuous systemic infusion in combination with cisplatin. Topical 5-FU demonstrated some efficacy, but in some cases viable tumor remained in the deeper dermis. The systemic chemotherapy resulted in one complete response and three partial responses in a total of five patients, suggesting that this therapy may be an option for treatment of extensive lesions. A dose reduction of 30% to 50% has been recommended for systemic chemotherapeutic agents in this population because of the increased sensitivity of XP cells.
Level of evidence: 3diii
For patients with EB, wide local excision of SCC with 2 cm margins remains the treatment of choice. Because the extent of SCC may be difficult to assess in these patients, clearance of SCC tumors may require continuous margin control surgery (Mohs excision) or generous clinical margins. Multiple surgical sessions may be required. Closure of the subsequent defects may require tissue rearrangement or dermal substitutes. Amputation may be considered as an option to reduce disease recurrence, although it is not clear that this has an impact on survival. The role of sentinel lymph node biopsy remains unclear in this population.
Current guidelines recommend that individuals with EB and unresectable SCC be treated with radiation therapy, but the dose may need to be given in smaller fractions in order to decrease the risk of skin desquamation. Systemic therapy with epidermal growth factor receptor antagonists or tyrosine kinase inhibitors may also be considered for individuals with advanced SCC.
Level of evidence: 5
For people who have genetic disorders other than XP, data are lacking, but general sun-safety measures remain important. Careful protection of the skin and eyes is the mainstay of prevention in all patients with increased susceptibility to skin cancer. Key points include avoidance of sun exposure at peak hours, protective clothing and lenses, and vigilant use of sunscreen. Avoidance of x-ray therapy has also been advocated for some groups with hereditary skin cancer syndromes, such as those with epidermodysplasia verruciformis. However, XP patients with unresectable skin cancers or internal cancers, such as spinal cord astrocytoma or glioblastomas of the brain, have been treated successfully with standard therapeutic doses of x-ray radiation. Some experts recommend dermatologic evaluation every 6 months and ophthalmologic evaluation at least annually in these high-risk populations. Guidelines for the management of patients with EB recommend skin examinations every 3 to 6 months starting at age 10 years for individuals with the RDEB-sev gen subtype of the disease. For individuals with other subtypes of EB, skin examination every 6 to 12 months starting at age 20 years is recommended in the absence of an established SCC diagnosis. Dental examination every 6 months is also recommended in this population.
Level of evidence: 5
For individuals with DEB, wound care is paramount. Use of silver sulfadiazine cream, medical grade honey, and soft silicone dressings can be helpful in these settings. Attention to nutritional status, which may be compromised because of esophageal strictures, iron-deficiency anemia, infection, and inflammation, is another critical consideration for wound healing for these patients. Multivitamin supplementation, often at higher doses than those routinely recommended for the general population, may be warranted.
Level of evidence: 5
Maintenance of skin integrity is a key factor for management of patients with JEB. A small study of five infants with severe generalized JEB with at least one premature truncating pathogenic variant in LAMB3 evaluated the clinical effects of treatment with gentamicin (7.5 mg/kg/day for 3 weeks) on skin fragility and other clinical features. Treatment did not impact overall mortality, but there was decreased skin fragility in four of the five patients; improved perceived quality of life as reported by caregivers, including increased physical activity without blistering; and increased laminin 332 expression in the skin.
Level of evidence: 3c
Bone marrow transplantation has been explored in patients with DEB; however, there is no evidence that this intervention results in a reduction of skin cancer. A double-blind, randomized, placebo-controlled trial of infusion of nonhematopoietic bone marrow stem cells with or without cyclosporine was conducted in 14 patients with recessive DEB. The rationale for this study was that mesenchymal stem cells (MSCs) have the potential to differentiate into dermal fibroblasts, the main expressor of type VII collagen. Seven subjects were randomly assigned to receive MSCs with 5 mg/kg/day of cyclosporine and an additional seven subjects received only MSCs. The number of new blisters and the rate of blister healing were significantly improved in both groups (P = .003 for the number of new blisters in the combination therapy group and P = .004 in the group receiving MSCs only; P< .001 for the rate of blister healing in both groups). However, no difference was seen between the groups.
Level of evidence (MSCs for blister prevention): 1b
Level of evidence (MSCs for blister treatment): 1
Future therapies for epidermolysis bullosa
Researchers are taking advantage of recent technological advances to study new strategies for the treatment of dominant and recessive EB. Clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9 (Cas9) is a technology that can be used to edit DNA. One research group used CRISPR/Cas9 to correct an inherited pathogenic variant in COL7A1 in keratinocytes isolated from a patient with RDEB. Keratinocytes that contained the corrected version of COL7A1 were successfully transplanted onto mice and staining of skin grafts after transplant showed normal skin. Another study used a different approach, retrovirus infection, to introduce normal COL7A1 into keratinocytes from four RDEB patients. The corrected keratinocytes were then assembled into epidermal graft sheets and transplanted onto six wound areas of each of the four patients. The grafts were well tolerated and showed greater healing capabilities than did noncorrected skin after further study. A small prospective trial of intravenous gentamicin in pediatric patients with junctional EB showed that patients had increased wound healing and improved quality of life after treatment. All of these therapies are still in early research stages and have not yet been evaluated in randomized, controlled clinical trials.
Level of evidence: None assigned
References
- Rogers HW, Weinstock MA, Harris AR, et al.: Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol 146 (3): 283-7, 2010.
- American Cancer Society: Cancer Facts and Figures 2023. American Cancer Society, 2023. Available online. Last accessed Dec. 15, 2023.
- Rogers HW, Weinstock MA, Feldman SR, et al.: Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the U.S. Population, 2012. JAMA Dermatol 151 (10): 1081-6, 2015.
- Flohil SC, van der Leest RJ, Arends LR, et al.: Risk of subsequent cutaneous malignancy in patients with prior keratinocyte carcinoma: a systematic review and meta-analysis. Eur J Cancer 49 (10): 2365-75, 2013.
- Armstrong BK, Kricker A: The epidemiology of UV induced skin cancer. J Photochem Photobiol B 63 (1-3): 8-18, 2001.
- Rosso S, Zanetti R, Martinez C, et al.: The multicentre south European study 'Helios'. II: Different sun exposure patterns in the aetiology of basal cell and squamous cell carcinomas of the skin. Br J Cancer 73 (11): 1447-54, 1996.
- Gallagher RP, Hill GB, Bajdik CD, et al.: Sunlight exposure, pigmentation factors, and risk of nonmelanocytic skin cancer. II. Squamous cell carcinoma. Arch Dermatol 131 (2): 164-9, 1995.
- Lindelöf B, Sigurgeirsson B, Tegner E, et al.: PUVA and cancer risk: the Swedish follow-up study. Br J Dermatol 141 (1): 108-12, 1999.
- Lim JL, Stern RS: High levels of ultraviolet B exposure increase the risk of non-melanoma skin cancer in psoralen and ultraviolet A-treated patients. J Invest Dermatol 124 (3): 505-13, 2005.
- Karagas MR, Stannard VA, Mott LA, et al.: Use of tanning devices and risk of basal cell and squamous cell skin cancers. J Natl Cancer Inst 94 (3): 224-6, 2002.
- Guo X, Fujino Y, Ye X, et al.: Association between multi-level inorganic arsenic exposure from drinking water and skin lesions in China. Int J Environ Res Public Health 3 (3): 262-7, 2006.
- Chen Y, Hall M, Graziano JH, et al.: A prospective study of blood selenium levels and the risk of arsenic-related premalignant skin lesions. Cancer Epidemiol Biomarkers Prev 16 (2): 207-13, 2007.
- Karagas MR, Stukel TA, Morris JS, et al.: Skin cancer risk in relation to toenail arsenic concentrations in a US population-based case-control study. Am J Epidemiol 153 (6): 559-65, 2001.
- Schwartz RA: Arsenic and the skin. Int J Dermatol 36 (4): 241-50, 1997.
- Mehta S, Goyal U, Gupta LK, et al.: Chronic Arsenicosis: Cases from a Nonendemic Area of South Rajasthan. Indian J Dermatol 64 (2): 164, 2019 Mar-Apr.
- Das KK, Chakaraborty A, Rahman A, et al.: Incidences of malignancy in chronic burn scar ulcers: experience from Bangladesh. Burns 41 (6): 1315-21, 2015.
- Koh D, Wang H, Lee J, et al.: Basal cell carcinoma, squamous cell carcinoma and melanoma of the skin: analysis of the Singapore Cancer Registry data 1968-97. Br J Dermatol 148 (6): 1161-6, 2003.
- Asgari MM, Warton EM, Whittemore AS: Family history of skin cancer is associated with increased risk of cutaneous squamous cell carcinoma. Dermatol Surg 41 (4): 481-6, 2015.
- Halder RM, Bang KM: Skin cancer in blacks in the United States. Dermatol Clin 6 (3): 397-405, 1988.
- Asuquo ME, Ebughe G: Major dermatological malignancies encountered in the University of Calabar Teaching Hospital, Calabar, southern Nigeria. Int J Dermatol 51 (Suppl 1): 32-6, 36-40, 2012.
- English DR, Armstrong BK, Kricker A, et al.: Demographic characteristics, pigmentary and cutaneous risk factors for squamous cell carcinoma of the skin: a case-control study. Int J Cancer 76 (5): 628-34, 1998.
- Kricker A, Armstrong BK, English DR, et al.: Pigmentary and cutaneous risk factors for non-melanocytic skin cancer--a case-control study. Int J Cancer 48 (5): 650-62, 1991.
- Akgüner M, Barutçu A, Yilmaz M, et al.: Marjolin's ulcer and chronic burn scarring. J Wound Care 7 (3): 121-2, 1998.
- Friedman R, Hanson S, Goldberg LH: Squamous cell carcinoma arising in a Leishmania scar. Dermatol Surg 29 (11): 1148-9, 2003.
- Jensen P, Hansen S, Møller B, et al.: Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol 40 (2 Pt 1): 177-86, 1999.
- Hartevelt MM, Bavinck JN, Kootte AM, et al.: Incidence of skin cancer after renal transplantation in The Netherlands. Transplantation 49 (3): 506-9, 1990.
- Lindelöf B, Sigurgeirsson B, Gäbel H, et al.: Incidence of skin cancer in 5356 patients following organ transplantation. Br J Dermatol 143 (3): 513-9, 2000.
- Krynitz B, Edgren G, Lindelöf B, et al.: Risk of skin cancer and other malignancies in kidney, liver, heart and lung transplant recipients 1970 to 2008--a Swedish population-based study. Int J Cancer 132 (6): 1429-38, 2013.
- Glover MT, Niranjan N, Kwan JT, et al.: Non-melanoma skin cancer in renal transplant recipients: the extent of the problem and a strategy for management. Br J Plast Surg 47 (2): 86-9, 1994.
- Kaplan AL, Cook JL: Cutaneous squamous cell carcinoma in patients with chronic lymphocytic leukemia. Skinmed 4 (5): 300-4, 2005 Sep-Oct.
- Euvrard S, Kanitakis J, Decullier E, et al.: Subsequent skin cancers in kidney and heart transplant recipients after the first squamous cell carcinoma. Transplantation 81 (8): 1093-100, 2006.
- Herrero JI, España A, D'Avola D, et al.: Subsequent nonmelanoma skin cancer after liver transplantation. Transplant Proc 44 (6): 1568-70, 2012 Jul-Aug.
- Cantwell MM, Murray LJ, Catney D, et al.: Second primary cancers in patients with skin cancer: a population-based study in Northern Ireland. Br J Cancer 100 (1): 174-7, 2009.
- Epstein E: Value of follow-up after treatment of basal cell carcinoma. Arch Dermatol 108 (6): 798-800, 1973.
- Møller R, Nielsen A, Reymann F: Multiple basal cell carcinoma and internal malignant tumors. Arch Dermatol 111 (5): 584-5, 1975.
- Bergstresser PR, Halprin KM: Multiple sequential skin cancers. The risk of skin cancer in patients with previous skin cancer. Arch Dermatol 111 (8): 995-6, 1975.
- Robinson JK: Risk of developing another basal cell carcinoma. A 5-year prospective study. Cancer 60 (1): 118-20, 1987.
- Greenberg ER, Baron JA, Stukel TA, et al.: A clinical trial of beta carotene to prevent basal-cell and squamous-cell cancers of the skin. The Skin Cancer Prevention Study Group. N Engl J Med 323 (12): 789-95, 1990.
- Karagas MR, Stukel TA, Greenberg ER, et al.: Risk of subsequent basal cell carcinoma and squamous cell carcinoma of the skin among patients with prior skin cancer. Skin Cancer Prevention Study Group. JAMA 267 (24): 3305-10, 1992.
- Helgadottir H, Höiom V, Tuominen R, et al.: CDKN2a mutation-negative melanoma families have increased risk exclusively for skin cancers but not for other malignancies. Int J Cancer 137 (9): 2220-6, 2015.
- Wei EX, Li X, Nan H: Having a first-degree relative with melanoma increases lifetime risk of melanoma, squamous cell carcinoma, and basal cell carcinoma. J Am Acad Dermatol 81 (2): 489-499, 2019.
- Hussain SK, Sundquist J, Hemminki K: The effect of having an affected parent or sibling on invasive and in situ skin cancer risk in Sweden. J Invest Dermatol 129 (9): 2142-7, 2009.
- Hemminki K, Zhang H, Czene K: Familial invasive and in situ squamous cell carcinoma of the skin. Br J Cancer 88 (9): 1375-80, 2003.
- Lindström LS, Yip B, Lichtenstein P, et al.: Etiology of familial aggregation in melanoma and squamous cell carcinoma of the skin. Cancer Epidemiol Biomarkers Prev 16 (8): 1639-43, 2007.
- Mucci LA, Hjelmborg JB, Harris JR, et al.: Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 315 (1): 68-76, 2016.
- Loh AYT, Špoljar S, Neo GYW, et al.: Huriez syndrome: Additional pathogenic variants supporting allelism to SMARCAD syndrome. Am J Med Genet A 188 (6): 1752-1760, 2022.
- DiGiovanna JJ, Kraemer KH: Shining a light on xeroderma pigmentosum. J Invest Dermatol 132 (3 Pt 2): 785-96, 2012.
- Kraemer KH, Lee MM, Andrews AD, et al.: The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Dermatol 130 (8): 1018-21, 1994.
- Bradford PT, Goldstein AM, Tamura D, et al.: Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet 48 (3): 168-76, 2011.
- Moussaid L, Benchikhi H, Boukind EH, et al.: [Cutaneous tumors during xeroderma pigmentosum in Morocco: study of 120 patients] Ann Dermatol Venereol 131 (1 Pt 1): 29-33, 2004.
- Brooks BP, Thompson AH, Bishop RJ, et al.: Ocular manifestations of xeroderma pigmentosum: long-term follow-up highlights the role of DNA repair in protection from sun damage. Ophthalmology 120 (7): 1324-36, 2013.
- Merideth M, Tamura D, Angra D, et al.: Reproductive Health in Xeroderma Pigmentosum: Features of Premature Aging. Obstet Gynecol 134 (4): 814-819, 2019.
- DiGiovanna JJ, Patronas N, Katz D, et al.: Xeroderma pigmentosum: spinal cord astrocytoma with 9-year survival after radiation and isotretinoin therapy. J Cutan Med Surg 2 (3): 153-8, 1998.
- Nikolaev S, Yurchenko AA, Sarasin A: Increased risk of internal tumors in DNA repair-deficient xeroderma pigmentosum patients: analysis of four international cohorts. Orphanet J Rare Dis 17 (1): 104, 2022.
- Robbins JH: Xeroderma pigmentosum complementation group H is withdrawn and reassigned to group D. Hum Genet 88 (2): 242, 1991.
- Khan SG, Oh KS, Shahlavi T, et al.: Reduced XPC DNA repair gene mRNA levels in clinically normal parents of xeroderma pigmentosum patients. Carcinogenesis 27 (1): 84-94, 2006.
- Hirai Y, Noda A, Kodama Y, et al.: Increased risk of skin cancer in Japanese heterozygotes of xeroderma pigmentosum group A. J Hum Genet 63 (11): 1181-1184, 2018.
- Messaoud O, Ben Rekaya M, Cherif W, et al.: Genetic homogeneity of mutational spectrum of group-A xeroderma pigmentosum in Tunisian patients. Int J Dermatol 49 (5): 544-8, 2010.
- Ben Rekaya M, Messaoud O, Talmoudi F, et al.: High frequency of the V548A fs X572 XPC mutation in Tunisia: implication for molecular diagnosis. J Hum Genet 54 (7): 426-9, 2009.
- Cartault F, Nava C, Malbrunot AC, et al.: A new XPC gene splicing mutation has lead to the highest worldwide prevalence of xeroderma pigmentosum in black Mahori patients. DNA Repair (Amst) 10 (6): 577-85, 2011.
- Doubaj Y, Laarabi FZ, Elalaoui SC, et al.: Carrier frequency of the recurrent mutation c.1643_1644delTG in the XPC gene and birth prevalence of the xeroderma pigmentosum in Morocco. J Dermatol 39 (4): 382-4, 2012.
- Hirai Y, Kodama Y, Moriwaki S, et al.: Heterozygous individuals bearing a founder mutation in the XPA DNA repair gene comprise nearly 1% of the Japanese population. Mutat Res 601 (1-2): 171-8, 2006.
- Vogelstein B, Knizler K: Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. In: Vogelstein B, Kinzler KW, eds.: The Genetic Basis of Human Cancer. 2nd ed. McGraw-Hill, 2002, pp 211-37.
- Kraemer KH, Slor H: Xeroderma pigmentosum. Clin Dermatol 3 (1): 33-69, 1985 Jan-Mar.
- Moriwaki S, Kraemer KH: Xeroderma pigmentosum--bridging a gap between clinic and laboratory. Photodermatol Photoimmunol Photomed 17 (2): 47-54, 2001.
- Ben Rekaya M, Laroussi N, Messaoud O, et al.: A founder large deletion mutation in Xeroderma pigmentosum-Variant form in Tunisia: implication for molecular diagnosis and therapy. Biomed Res Int 2014: 256245, 2014.
- Fassihi H, Sethi M, Fawcett H, et al.: Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect. Proc Natl Acad Sci U S A 113 (9): E1236-45, 2016.
- Amr K, Messaoud O, El Darouti M, et al.: Mutational spectrum of Xeroderma pigmentosum group A in Egyptian patients. Gene 533 (1): 52-6, 2014.
- Schriver C, Cleaver J, et al., eds.: Xeroderma pigmentosum and cockayne syndrome. In: Cleaver J, Kraemer K, eds.: The Metabolic and Molecular Bases of Inherited Disease. 7th ed. McGraw-Hill Book Co, 1995, pp 4397.
- Lambert WC, Gagna CE, Lambert MW: Xeroderma pigmentosum: its overlap with trichothiodystrophy, Cockayne syndrome and other progeroid syndromes. Adv Exp Med Biol 637: 128-37, 2008.
- Robbins JH, Kraemer KH, Lutzner MA, et al.: Xeroderma pigmentosum. An inherited diseases with sun sensitivity, multiple cutaneous neoplasms, and abnormal DNA repair. Ann Intern Med 80 (2): 221-48, 1974.
- Weeda G, Eveno E, Donker I, et al.: A mutation in the XPB/ERCC3 DNA repair transcription gene, associated with trichothiodystrophy. Am J Hum Genet 60 (2): 320-9, 1997.
- Lehmann AR: The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes Dev 15 (1): 15-23, 2001.
- Broughton BC, Berneburg M, Fawcett H, et al.: Two individuals with features of both xeroderma pigmentosum and trichothiodystrophy highlight the complexity of the clinical outcomes of mutations in the XPD gene. Hum Mol Genet 10 (22): 2539-47, 2001.
- Goudie DR, Yuille MA, Leversha MA, et al.: Multiple self-healing squamous epitheliomata (ESS1) mapped to chromosome 9q22-q31 in families with common ancestry. Nat Genet 3 (2): 165-9, 1993.
- Goudie DR, D'Alessandro M, Merriman B, et al.: Multiple self-healing squamous epithelioma is caused by a disease-specific spectrum of mutations in TGFBR1. Nat Genet 43 (4): 365-9, 2011.
- Bose S, Morgan LJ, Booth DR, et al.: The elusive multiple self-healing squamous epithelioma (MSSE) gene: further mapping, analysis of candidates, and loss of heterozygosity. Oncogene 25 (5): 806-12, 2006.
- Mabula JB, Chalya PL, Mchembe MD, et al.: Skin cancers among Albinos at a University teaching hospital in Northwestern Tanzania: a retrospective review of 64 cases. BMC Dermatol 12: 5, 2012.
- Luande J, Henschke CI, Mohammed N: The Tanzanian human albino skin. Natural history. Cancer 55 (8): 1823-8, 1985.
- Knöpfel N, Martín-Santiago A, Del Pozo LJ, et al.: Amelanotic naevoid melanoma in a 16-month-old albino infant. Clin Exp Dermatol 42 (1): 84-88, 2017.
- Iversen U, Iversen OH: Tumours of the skin. In: Templeton AC, ed.: Tumours in a Tropical Country: A Survey of Uganda, 1964-1968. Springer, 1973, pp 180-99.
- Hutton SM, Spritz RA: A comprehensive genetic study of autosomal recessive ocular albinism in Caucasian patients. Invest Ophthalmol Vis Sci 49 (3): 868-72, 2008.
- Laetitia MM, Veronique K, Mamy NZ, et al.: Molecular genetic characterization of Congolese patients with oculocutaneous albinism. Eur J Med Genet 65 (11): 104611, 2022.
- Brilliant MH: The mouse p (pink-eyed dilution) and human P genes, oculocutaneous albinism type 2 (OCA2), and melanosomal pH. Pigment Cell Res 14 (2): 86-93, 2001.
- Sviderskaya EV, Bennett DC, Ho L, et al.: Complementation of hypopigmentation in p-mutant (pink-eyed dilution) mouse melanocytes by normal human P cDNA, and defective complementation by OCA2 mutant sequences. J Invest Dermatol 108 (1): 30-4, 1997.
- Mauri L, Manfredini E, Del Longo A, et al.: Clinical evaluation and molecular screening of a large consecutive series of albino patients. J Hum Genet 62 (2): 277-290, 2017.
- Inagaki K, Suzuki T, Shimizu H, et al.: Oculocutaneous albinism type 4 is one of the most common types of albinism in Japan. Am J Hum Genet 74 (3): 466-71, 2004.
- Mauri L, Barone L, Al Oum M, et al.: SLC45A2 mutation frequency in Oculocutaneous Albinism Italian patients doesn't differ from other European studies. Gene 533 (1): 398-402, 2014.
- Simeonov DR, Wang X, Wang C, et al.: DNA variations in oculocutaneous albinism: an updated mutation list and current outstanding issues in molecular diagnostics. Hum Mutat 34 (6): 827-35, 2013.
- Moreno-Artero E, Morice-Picard F, Lasseaux E, et al.: Oculo-Cutaneous Albinism Type 4 (OCA4): Phenotype-Genotype Correlation. Genes (Basel) 13 (12): , 2022.
- Ibarrola-Villava M, Hu HH, Guedj M, et al.: MC1R, SLC45A2 and TYR genetic variants involved in melanoma susceptibility in southern European populations: results from a meta-analysis. Eur J Cancer 48 (14): 2183-91, 2012.
- Kausar T, Bhatti MA, Ali M, et al.: OCA5, a novel locus for non-syndromic oculocutaneous albinism, maps to chromosome 4q24. Clin Genet 84 (1): 91-3, 2013.
- Wei AH, Zang DJ, Zhang Z, et al.: Exome sequencing identifies SLC24A5 as a candidate gene for nonsyndromic oculocutaneous albinism. J Invest Dermatol 133 (7): 1834-40, 2013.
- Morice-Picard F, Lasseaux E, François S, et al.: SLC24A5 mutations are associated with non-syndromic oculocutaneous albinism. J Invest Dermatol 134 (2): 568-571, 2014.
- Grønskov K, Dooley CM, Østergaard E, et al.: Mutations in c10orf11, a melanocyte-differentiation gene, cause autosomal-recessive albinism. Am J Hum Genet 92 (3): 415-21, 2013.
- Nathan V, Johansson PA, Palmer JM, et al.: Germline variants in oculocutaneous albinism genes and predisposition to familial cutaneous melanoma. Pigment Cell Melanoma Res 32 (6): 854-863, 2019.
- Tomita Y, Miyamura Y: Oculocutaneous albinism and analysis of tyrosinase gene in Japanese patients. Nagoya J Med Sci 61 (3-4): 97-102, 1998.
- Liu N, Kong XD, Shi HR, et al.: Tyrosinase gene mutations in the Chinese Han population with OCA1. Genet Res (Camb) 96: e14, 2014.
- FROGGATT P: Albinism in Northern Ireland. Ann Hum Genet 24: 213-38, 1960.
- McLeod R, Lowry RB: Incidence of albinism in British Columbia (B.C.). Separation by hairbulb test. Clin Genet 9 (1): 77-80, 1976.
- Martínez-Frías ML, Bermejo E: Prevalence of congenital anomaly syndromes in a Spanish gypsy population. J Med Genet 29 (7): 483-6, 1992.
- Grønskov K, Ek J, Sand A, et al.: Birth prevalence and mutation spectrum in danish patients with autosomal recessive albinism. Invest Ophthalmol Vis Sci 50 (3): 1058-64, 2009.
- Chiang PW, Drautz JM, Tsai AC, et al.: A new hypothesis of OCA1B. Am J Med Genet A 146A (22): 2968-70, 2008.
- Okoro AN: Albinism in Nigeria. A clinical and social study. Br J Dermatol 92 (5): 485-92, 1975.
- Kagore F, Lund PM: Oculocutaneous albinism among schoolchildren in Harare, Zimbabwe. J Med Genet 32 (11): 859-61, 1995.
- Lee ST, Nicholls RD, Bundey S, et al.: Mutations of the P gene in oculocutaneous albinism, ocular albinism, and Prader-Willi syndrome plus albinism. N Engl J Med 330 (8): 529-34, 1994.
- WOOLF CM: ALBINISM AMONG INDIANS IN ARIZONA AND NEW MEXICO. Am J Hum Genet 17: 23-35, 1965.
- Manga P, Kromberg JG, Box NF, et al.: Rufous oculocutaneous albinism in southern African Blacks is caused by mutations in the TYRP1 gene. Am J Hum Genet 61 (5): 1095-101, 1997.
- Rundshagen U, Zühlke C, Opitz S, et al.: Mutations in the MATP gene in five German patients affected by oculocutaneous albinism type 4. Hum Mutat 23 (2): 106-10, 2004.
- Bertolotti A, Lasseaux E, Plaisant C, et al.: Identification of a homozygous mutation of SLC24A5 (OCA6) in two patients with oculocutaneous albinism from French Guiana. Pigment Cell Melanoma Res 29 (1): 104-6, 2016.
- Mondal M, Sengupta M, Samanta S, et al.: Molecular basis of albinism in India: evaluation of seven potential candidate genes and some new findings. Gene 511 (2): 470-4, 2012.
- Perry PK, Silverberg NB: Cutaneous malignancy in albinism. Cutis 67 (5): 427-30, 2001.
- Fukai K, Oh J, Frenk E, et al.: Linkage disequilibrium mapping of the gene for Hermansky-Pudlak syndrome to chromosome 10q23.1-q23.3. Hum Mol Genet 4 (9): 1665-9, 1995.
- Wildenberg SC, Oetting WS, Almodóvar C, et al.: A gene causing Hermansky-Pudlak syndrome in a Puerto Rican population maps to chromosome 10q2. Am J Hum Genet 57 (4): 755-65, 1995.
- Anikster Y, Huizing M, White J, et al.: Mutation of a new gene causes a unique form of Hermansky-Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat Genet 28 (4): 376-80, 2001.
- Suzuki T, Li W, Zhang Q, et al.: Hermansky-Pudlak syndrome is caused by mutations in HPS4, the human homolog of the mouse light-ear gene. Nat Genet 30 (3): 321-4, 2002.
- Zhang Q, Zhao B, Li W, et al.: Ru2 and Ru encode mouse orthologs of the genes mutated in human Hermansky-Pudlak syndrome types 5 and 6. Nat Genet 33 (2): 145-53, 2003.
- Li W, Zhang Q, Oiso N, et al.: Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat Genet 35 (1): 84-9, 2003.
- Morgan NV, Pasha S, Johnson CA, et al.: A germline mutation in BLOC1S3/reduced pigmentation causes a novel variant of Hermansky-Pudlak syndrome (HPS8). Am J Hum Genet 78 (1): 160-6, 2006.
- Cullinane AR, Curry JA, Carmona-Rivera C, et al.: A BLOC-1 mutation screen reveals that PLDN is mutated in Hermansky-Pudlak Syndrome type 9. Am J Hum Genet 88 (6): 778-87, 2011.
- Toro J, Turner M, Gahl WA: Dermatologic manifestations of Hermansky-Pudlak syndrome in patients with and without a 16-base pair duplication in the HPS1 gene. Arch Dermatol 135 (7): 774-80, 1999.
- Dell'Angelica EC, Shotelersuk V, Aguilar RC, et al.: Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell 3 (1): 11-21, 1999.
- Nagle DL, Karim MA, Woolf EA, et al.: Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet 14 (3): 307-11, 1996.
- Perou CM, Moore KJ, Nagle DL, et al.: Identification of the murine beige gene by YAC complementation and positional cloning. Nat Genet 13 (3): 303-8, 1996.
- Barbosa MD, Nguyen QA, Tchernev VT, et al.: Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature 382 (6588): 262-5, 1996.
- Engle LJ, Kennett RH: Cloning, analysis, and chromosomal localization of myoxin (MYH12), the human homologue to the mouse dilute gene. Genomics 19 (3): 407-16, 1994.
- Ménasché G, Pastural E, Feldmann J, et al.: Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet 25 (2): 173-6, 2000.
- Ménasché G, Ho CH, Sanal O, et al.: Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest 112 (3): 450-6, 2003.
- Baardman R, Yenamandra VK, Duipmans JC, et al.: Novel insights into the epidemiology of epidermolysis bullosa (EB) from the Dutch EB Registry: EB more common than previously assumed? J Eur Acad Dermatol Venereol 35 (4): 995-1006, 2021.
- Has C, Hess M, Anemüller W, et al.: Epidemiology of inherited epidermolysis bullosa in Germany. J Eur Acad Dermatol Venereol 37 (2): 402-410, 2023.
- Petrof G, Papanikolaou M, Martinez AE, et al.: The epidemiology of epidermolysis bullosa in England and Wales: data from the national epidermolysis bullosa database. Br J Dermatol 186 (5): 843-848, 2022.
- Mariath LM, Santin JT, Frantz JA, et al.: Genotype-phenotype correlations on epidermolysis bullosa with congenital absence of skin: A comprehensive review. Clin Genet 99 (1): 29-41, 2021.
- Montaudié H, Chiaverini C, Sbidian E, et al.: Inherited epidermolysis bullosa and squamous cell carcinoma: a systematic review of 117 cases. Orphanet J Rare Dis 11 (1): 117, 2016.
- Phillips GS, Huang A, Augsburger BD, et al.: A retrospective analysis of diagnostic testing in a large North American cohort of patients with epidermolysis bullosa. J Am Acad Dermatol 86 (5): 1063-1071, 2022.
- Rossi S, Castiglia D, Pisaneschi E, et al.: Immunofluorescence mapping, electron microscopy and genetics in the diagnosis and sub-classification of inherited epidermolysis bullosa: a single-centre retrospective comparative study of 87 cases with long-term follow-up. J Eur Acad Dermatol Venereol 35 (4): 1007-1016, 2021.
- Has C, Bauer JW, Bodemer C, et al.: Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol 183 (4): 614-627, 2020.
- Bruckner-Tuderman L: Hereditary skin diseases of anchoring fibrils. J Dermatol Sci 20 (2): 122-33, 1999.
- van den Akker PC, Jonkman MF, Rengaw T, et al.: The international dystrophic epidermolysis bullosa patient registry: an online database of dystrophic epidermolysis bullosa patients and their COL7A1 mutations. Hum Mutat 32 (10): 1100-7, 2011.
- Fine JD, Bruckner-Tuderman L, Eady RA, et al.: Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 70 (6): 1103-26, 2014.
- Fine JD: Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates From the National Epidermolysis Bullosa Registry. JAMA Dermatol 152 (11): 1231-1238, 2016.
- Fine J, Johnson L, Suchindran C, et al.: Cancer and inherited epidermolysis bullosa. In: Fine J, Bauer E, McGuire J, et al., eds.: Epidermolysis Bullosa; Clinical, Epidemiologic, and Laboratory Advances and the Findings of the National Epidermolysis Bullosa Registry. The Johns Hopkins University Press, 1999, pp 175-92.
- Fine JD, Johnson LB, Weiner M, et al.: Chemoprevention of squamous cell carcinoma in recessive dystrophic epidermolysis bullosa: results of a phase 1 trial of systemic isotretinoin. J Am Acad Dermatol 50 (4): 563-71, 2004.
- Fine JD, Johnson LB, Weiner M, et al.: Cause-specific risks of childhood death in inherited epidermolysis bullosa. J Pediatr 152 (2): 276-80, 2008.
- van den Akker PC, van Essen AJ, Kraak MM, et al.: Long-term follow-up of patients with recessive dystrophic epidermolysis bullosa in the Netherlands: expansion of the mutation database and unusual phenotype-genotype correlations. J Dermatol Sci 56 (1): 9-18, 2009.
- Feinstein JA, Jambal P, Peoples K, et al.: Assessment of the Timing of Milestone Clinical Events in Patients With Epidermolysis Bullosa From North America. JAMA Dermatol 155 (2): 196-203, 2019.
- Farhi D: Surgical management of epidermolysis bullosa: the importance of a multidisciplinary management. Int J Dermatol 46 (8): 815-6, 2007.
- Wessagowit V, Ashton GH, Mohammedi R, et al.: Three cases of de novo dominant dystrophic epidermolysis bullosa associated with the mutation G2043R in COL7A1. Clin Exp Dermatol 26 (1): 97-9, 2001.
- Cserhalmi-Friedman PB, Garzon MC, Guzman E, et al.: Maternal germline mosaicism in dominant dystrophic epidermolysis bullosa. J Invest Dermatol 117 (5): 1327-8, 2001.
- Cuadrado-Corrales N, Sánchez-Jimeno C, García M, et al.: A prevalent mutation with founder effect in Spanish Recessive Dystrophic Epidermolysis Bullosa families. BMC Med Genet 11: 139, 2010.
- Ben Brick AS, Laroussi N, Mesrati H, et al.: Mutational founder effect in recessive dystrophic epidermolysis bullosa families from Southern Tunisia. Arch Dermatol Res 306 (4): 405-11, 2014.
- Ortiz-Urda S, Garcia J, Green CL, et al.: Type VII collagen is required for Ras-driven human epidermal tumorigenesis. Science 307 (5716): 1773-6, 2005.
- Fine JD: Inherited epidermolysis bullosa. Orphanet J Rare Dis 5: 12, 2010.
- Aumailley M, Bruckner-Tuderman L, Carter WG, et al.: A simplified laminin nomenclature. Matrix Biol 24 (5): 326-32, 2005.
- Nakano A, Chao SC, Pulkkinen L, et al.: Laminin 5 mutations in junctional epidermolysis bullosa: molecular basis of Herlitz vs. non-Herlitz phenotypes. Hum Genet 110 (1): 41-51, 2002.
- Schumann H, Hammami-Hauasli N, Pulkkinen L, et al.: Three novel homozygous point mutations and a new polymorphism in the COL17A1 gene: relation to biological and clinical phenotypes of junctional epidermolysis bullosa. Am J Hum Genet 60 (6): 1344-53, 1997.
- Fine JD, Mellerio JE: Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. J Am Acad Dermatol 61 (3): 367-84; quiz 385-6, 2009.
- Kiritsi D, Kern JS, Schumann H, et al.: Molecular mechanisms of phenotypic variability in junctional epidermolysis bullosa. J Med Genet 48 (7): 450-7, 2011.
- Majewski S, Jabłońska S: Epidermodysplasia verruciformis as a model of human papillomavirus-induced genetic cancer of the skin. Arch Dermatol 131 (11): 1312-8, 1995.
- Sterling JC: Human papillomaviruses and skin cancer. J Clin Virol 32 (Suppl 1): S67-71, 2005.
- Karagas MR, Nelson HH, Sehr P, et al.: Human papillomavirus infection and incidence of squamous cell and basal cell carcinomas of the skin. J Natl Cancer Inst 98 (6): 389-95, 2006.
- Ramoz N, Rueda LA, Bouadjar B, et al.: Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat Genet 32 (4): 579-81, 2002.
- Mulvihill J, Miller R, Fraumeni J, eds.: Nosology among the neoplastic genedermatoses. In: Mulvihill J, Miller R, Fraumeni J, eds.: Genetics of Human Cancer. Raven Press, 1977, pp 145-67.
- Jabłońska S, Orth G, Jarzabek-Chorzelska M, et al.: Twenty-one years of follow-up studies of familial epidermodysplasia verruciformis. Dermatologica 158 (5): 309-27, 1979.
- McDermott DF, Gammon B, Snijders PJ, et al.: Autosomal dominant epidermodysplasia verruciformis lacking a known EVER1 or EVER2 mutation. Pediatr Dermatol 26 (3): 306-10, 2009 May-Jun.
- Lazarczyk M, Pons C, Mendoza JA, et al.: Regulation of cellular zinc balance as a potential mechanism of EVER-mediated protection against pathogenesis by cutaneous oncogenic human papillomaviruses. J Exp Med 205 (1): 35-42, 2008.
- Patel AS, Karagas MR, Pawlita M, et al.: Cutaneous human papillomavirus infection, the EVER2 gene and incidence of squamous cell carcinoma: a case-control study. Int J Cancer 122 (10): 2377-9, 2008.
- Crequer A, Picard C, Patin E, et al.: Inherited MST1 deficiency underlies susceptibility to EV-HPV infections. PLoS One 7 (8): e44010, 2012.
- Ramoz N, Taïeb A, Rueda LA, et al.: Evidence for a nonallelic heterogeneity of epidermodysplasia verruciformis with two susceptibility loci mapped to chromosome regions 2p21-p24 and 17q25. J Invest Dermatol 114 (6): 1148-53, 2000.
- Rosenberg PS, Tamary H, Alter BP: How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi Anemia in the United States and Israel. Am J Med Genet A 155A (8): 1877-83, 2011.
- Rosenberg PS, Greene MH, Alter BP: Cancer incidence in persons with Fanconi anemia. Blood 101 (3): 822-6, 2003.
- Alter BP: Cancer in Fanconi anemia, 1927-2001. Cancer 97 (2): 425-40, 2003.
- Puligandla B, Stass SA, Schumacher HR, et al.: Terminal deoxynucleotidyl transferase in Fanconi's anaemia. Lancet 2 (8102): 1263, 1978.
- Alter BP, Frissora CL, Halpérin DS, et al.: Fanconi's anaemia and pregnancy. Br J Haematol 77 (3): 410-8, 1991.
- Berger R, Le Coniat M, Schaison G: Chromosome abnormalities in bone marrow of Fanconi anemia patients. Cancer Genet Cytogenet 65 (1): 47-50, 1993.
- Lebbé C, Pinquier L, Rybojad M, et al.: Fanconi's anaemia associated with multicentric Bowen's disease and decreased NK cytotoxicity. Br J Dermatol 129 (5): 615-8, 1993.
- Bagby GC, Alter BP: Fanconi anemia. Semin Hematol 43 (3): 147-56, 2006.
- Chang L, Yuan W, Zeng H, et al.: Whole exome sequencing reveals concomitant mutations of multiple FA genes in individual Fanconi anemia patients. BMC Med Genomics 7: 24, 2014.
- Wang W: Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet 8 (10): 735-48, 2007.
- Howlett NG, Taniguchi T, Olson S, et al.: Biallelic inactivation of BRCA2 in Fanconi anemia. Science 297 (5581): 606-9, 2002.
- Seal S, Thompson D, Renwick A, et al.: Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet 38 (11): 1239-41, 2006.
- Berwick M, Satagopan JM, Ben-Porat L, et al.: Genetic heterogeneity among Fanconi anemia heterozygotes and risk of cancer. Cancer Res 67 (19): 9591-6, 2007.
- Rafnar T, Sigurjonsdottir GR, Stacey SN, et al.: Association of BRCA2 K3326* With Small Cell Lung Cancer and Squamous Cell Cancer of the Skin. J Natl Cancer Inst 110 (9): 967-974, 2018.
- Walne AJ, Vulliamy T, Beswick R, et al.: TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood 112 (9): 3594-600, 2008.
- Alter BP, Giri N, Savage SA, et al.: Cancer in dyskeratosis congenita. Blood 113 (26): 6549-57, 2009.
- Vulliamy T, Dokal I: Dyskeratosis congenita. Semin Hematol 43 (3): 157-66, 2006.
- Knight SW, Heiss NS, Vulliamy TJ, et al.: X-linked dyskeratosis congenita is predominantly caused by missense mutations in the DKC1 gene. Am J Hum Genet 65 (1): 50-8, 1999.
- Vulliamy T, Marrone A, Szydlo R, et al.: Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat Genet 36 (5): 447-9, 2004.
- Vulliamy TJ, Walne A, Baskaradas A, et al.: Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure. Blood Cells Mol Dis 34 (3): 257-63, 2005 May-Jun.
- Vulliamy T, Beswick R, Kirwan M, et al.: Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc Natl Acad Sci U S A 105 (23): 8073-8, 2008.
- Walne AJ, Vulliamy T, Marrone A, et al.: Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet 16 (13): 1619-29, 2007.
- Savage SA, Giri N, Baerlocher GM, et al.: TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet 82 (2): 501-9, 2008.
- Marrone A, Walne A, Tamary H, et al.: Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood 110 (13): 4198-205, 2007.
- Ballew BJ, Yeager M, Jacobs K, et al.: Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum Genet 132 (4): 473-80, 2013.
- Walne AJ, Vulliamy T, Kirwan M, et al.: Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet 92 (3): 448-53, 2013.
- Fedick AM, Shi L, Jalas C, et al.: Carrier screening of RTEL1 mutations in the Ashkenazi Jewish population. Clin Genet 88 (2): 177-81, 2015.
- Batista LF, Pech MF, Zhong FL, et al.: Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature 474 (7351): 399-402, 2011.
- Neveling K, Bechtold A, Hoehn H: Genetic instability syndromes with progeroid features. Z Gerontol Geriatr 40 (5): 339-48, 2007.
- Zhong F, Savage SA, Shkreli M, et al.: Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev 25 (1): 11-6, 2011.
- Walne AJ, Vulliamy T, Beswick R, et al.: Mutations in C16orf57 and normal-length telomeres unify a subset of patients with dyskeratosis congenita, poikiloderma with neutropenia and Rothmund-Thomson syndrome. Hum Mol Genet 19 (22): 4453-61, 2010.
- Colombo EA, Bazan JF, Negri G, et al.: Novel C16orf57 mutations in patients with Poikiloderma with Neutropenia: bioinformatic analysis of the protein and predicted effects of all reported mutations. Orphanet J Rare Dis 7: 7, 2012.
- Alter BP, Baerlocher GM, Savage SA, et al.: Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood 110 (5): 1439-47, 2007.
- Vulliamy TJ, Marrone A, Knight SW, et al.: Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood 107 (7): 2680-5, 2006.
- Vulliamy TJ, Dokal I: Dyskeratosis congenita: the diverse clinical presentation of mutations in the telomerase complex. Biochimie 90 (1): 122-30, 2008.
- Borg MF, Olver IN, Hill MP: Rothmund-Thomson syndrome and tolerance of chemoradiotherapy. Australas Radiol 42 (3): 216-8, 1998.
- Haneke E, Gutschmidt E: Premature multiple Bowen's disease in poikiloderma congenitale with warty hyperkeratoses. Dermatologica 158 (5): 384-8, 1979.
- Wang LL, Levy ML, Lewis RA, et al.: Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am J Med Genet 102 (1): 11-7, 2001.
- Piquero-Casals J, Okubo AY, Nico MM: Rothmund-thomson syndrome in three siblings and development of cutaneous squamous cell carcinoma. Pediatr Dermatol 19 (4): 312-6, 2002 Jul-Aug.
- Petkovic M, Dietschy T, Freire R, et al.: The human Rothmund-Thomson syndrome gene product, RECQL4, localizes to distinct nuclear foci that coincide with proteins involved in the maintenance of genome stability. J Cell Sci 118 (Pt 18): 4261-9, 2005.
- Wang LL, Gannavarapu A, Kozinetz CA, et al.: Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst 95 (9): 669-74, 2003.
- Kitao S, Lindor NM, Shiratori M, et al.: Rothmund-thomson syndrome responsible gene, RECQL4: genomic structure and products. Genomics 61 (3): 268-76, 1999.
- Werner SR, Prahalad AK, Yang J, et al.: RECQL4-deficient cells are hypersensitive to oxidative stress/damage: Insights for osteosarcoma prevalence and heterogeneity in Rothmund-Thomson syndrome. Biochem Biophys Res Commun 345 (1): 403-9, 2006.
- Nakayama H: RecQ family helicases: roles as tumor suppressor proteins. Oncogene 21 (58): 9008-21, 2002.
- Ajeawung NF, Nguyen TTM, Lu L, et al.: Mutations in ANAPC1, Encoding a Scaffold Subunit of the Anaphase-Promoting Complex, Cause Rothmund-Thomson Syndrome Type 1. Am J Hum Genet 105 (3): 625-630, 2019.
- Sugrañes TA, Flanagan M, Thomas C, et al.: Age of first cancer diagnosis and survival in Bloom syndrome. Genet Med 24 (7): 1476-1484, 2022.
- Cunniff C, Djavid AR, Carrubba S, et al.: Health supervision for people with Bloom syndrome. Am J Med Genet A 176 (9): 1872-1881, 2018.
- Ellis NA, Groden J, Ye TZ, et al.: The Bloom's syndrome gene product is homologous to RecQ helicases. Cell 83 (4): 655-66, 1995.
- Bugreev DV, Yu X, Egelman EH, et al.: Novel pro- and anti-recombination activities of the Bloom's syndrome helicase. Genes Dev 21 (23): 3085-94, 2007.
- German J, Ellis N: Bloom syndrome. In: Vogelstein B, Kinzler KW, eds.: The Genetic Basis of Human Cancer. 2nd ed. McGraw-Hill, 2002, pp 267-88.
- German J, Sanz MM, Ciocci S, et al.: Syndrome-causing mutations of the BLM gene in persons in the Bloom's Syndrome Registry. Hum Mutat 28 (8): 743-53, 2007.
- Ellis NA, Ciocci S, Proytcheva M, et al.: The Ashkenazic Jewish Bloom syndrome mutation blmAsh is present in non-Jewish Americans of Spanish ancestry. Am J Hum Genet 63 (6): 1685-93, 1998.
- Lu X, Lane DP: Differential induction of transcriptionally active p53 following UV or ionizing radiation: defects in chromosome instability syndromes? Cell 75 (4): 765-78, 1993.
- Meetei AR, Sechi S, Wallisch M, et al.: A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol 23 (10): 3417-26, 2003.
- Yamamoto K, Imakiire A, Miyagawa N, et al.: A report of two cases of Werner's syndrome and review of the literature. J Orthop Surg (Hong Kong) 11 (2): 224-33, 2003.
- Huang S, Lee L, Hanson NB, et al.: The spectrum of WRN mutations in Werner syndrome patients. Hum Mutat 27 (6): 558-67, 2006.
- Goto M, Miller RW, Ishikawa Y, et al.: Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev 5 (4): 239-46, 1996.
- Tsuchiya H, Tomita K, Ohno M, et al.: Werner's syndrome combined with quintuplicate malignant tumors: a case report and review of literature data. Jpn J Clin Oncol 21 (2): 135-42, 1991.
- Lauper JM, Krause A, Vaughan TL, et al.: Spectrum and risk of neoplasia in Werner syndrome: a systematic review. PLoS One 8 (4): e59709, 2013.
- Machino H, Miki Y, Teramoto T, et al.: Cytogenetic studies in a patient with porokeratosis of Mibelli, multiple cancers and a forme fruste of Werner's syndrome. Br J Dermatol 111 (5): 579-86, 1984.
- Goto M, Imamura O, Kuromitsu J, et al.: Analysis of helicase gene mutations in Japanese Werner's syndrome patients. Hum Genet 99 (2): 191-3, 1997.
- Oshima J, Yu CE, Piussan C, et al.: Homozygous and compound heterozygous mutations at the Werner syndrome locus. Hum Mol Genet 5 (12): 1909-13, 1996.
- Uhrhammer NA, Lafarge L, Dos Santos L, et al.: Werner syndrome and mutations of the WRN and LMNA genes in France. Hum Mutat 27 (7): 718-9, 2006.
- Yu CE, Oshima J, Wijsman EM, et al.: Mutations in the consensus helicase domains of the Werner syndrome gene. Werner's Syndrome Collaborative Group. Am J Hum Genet 60 (2): 330-41, 1997.
- Shen JC, Loeb LA: The Werner syndrome gene: the molecular basis of RecQ helicase-deficiency diseases. Trends Genet 16 (5): 213-20, 2000.
- Shen J, Loeb LA: Unwinding the molecular basis of the Werner syndrome. Mech Ageing Dev 122 (9): 921-44, 2001.
- Brosh RM, Bohr VA: Roles of the Werner syndrome protein in pathways required for maintenance of genome stability. Exp Gerontol 37 (4): 491-506, 2002.
- Furuichi Y: Premature aging and predisposition to cancers caused by mutations in RecQ family helicases. Ann N Y Acad Sci 928: 121-31, 2001.
- Lebel M: Werner syndrome: genetic and molecular basis of a premature aging disorder. Cell Mol Life Sci 58 (7): 857-67, 2001.
- Bohr VA, Brosh RM, von Kobbe C, et al.: Pathways defective in the human premature aging disease Werner syndrome. Biogerontology 3 (1-2): 89-94, 2002.
- Chen L, Oshima J: Werner Syndrome. J Biomed Biotechnol 2 (2): 46-54, 2002.
- Opresko PL, Cheng WH, von Kobbe C, et al.: Werner syndrome and the function of the Werner protein; what they can teach us about the molecular aging process. Carcinogenesis 24 (5): 791-802, 2003.
- Pirzio LM, Pichierri P, Bignami M, et al.: Werner syndrome helicase activity is essential in maintaining fragile site stability. J Cell Biol 180 (2): 305-14, 2008.
- Crabbe L, Jauch A, Naeger CM, et al.: Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc Natl Acad Sci U S A 104 (7): 2205-10, 2007.
- Friedrich K, Lee L, Leistritz DF, et al.: WRN mutations in Werner syndrome patients: genomic rearrangements, unusual intronic mutations and ethnic-specific alterations. Hum Genet 128 (1): 103-11, 2010.
- Matsumoto T, Imamura O, Yamabe Y, et al.: Mutation and haplotype analyses of the Werner's syndrome gene based on its genomic structure: genetic epidemiology in the Japanese population. Hum Genet 100 (1): 123-30, 1997.
- Satoh M, Imai M, Sugimoto M, et al.: Prevalence of Werner's syndrome heterozygotes in Japan. Lancet 353 (9166): 1766, 1999.
- Moser MJ, Oshima J, Monnat RJ: WRN mutations in Werner syndrome. Hum Mutat 13 (4): 271-9, 1999.
- Goto M, Yamabe Y, Shiratori M, et al.: Immunological diagnosis of Werner syndrome by down-regulated and truncated gene products. Hum Genet 105 (4): 301-7, 1999.
- Tagliabue E, Fargnoli MC, Gandini S, et al.: MC1R gene variants and non-melanoma skin cancer: a pooled-analysis from the M-SKIP project. Br J Cancer 113 (2): 354-63, 2015.
- Ferrucci LM, Cartmel B, Molinaro AM, et al.: Host phenotype characteristics and MC1R in relation to early-onset basal cell carcinoma. J Invest Dermatol 132 (4): 1272-9, 2012.
- Chen AC, Martin AJ, Choy B, et al.: A Phase 3 Randomized Trial of Nicotinamide for Skin-Cancer Chemoprevention. N Engl J Med 373 (17): 1618-26, 2015.
- Tamura D, DiGiovanna JJ, Khan SG, et al.: Living with xeroderma pigmentosum: comprehensive photoprotection for highly photosensitive patients. Photodermatol Photoimmunol Photomed 30 (2-3): 146-52, 2014 Apr-Jun.
- Tamura D, DiGiovanna JJ, Kraemer KH: Xeroderma pigmentosum. In: Lebwohl MG, Birth-Jones J, Heymann WR, et al., eds.: Treatment of Skin Disease: Comprehensive Therapeutic Strategies. 3rd ed. Saunders Elsevier, 2010, pp 789-92.
- Kraemer KH, DiGiovanna JJ, Moshell AN, et al.: Prevention of skin cancer in xeroderma pigmentosum with the use of oral isotretinoin. N Engl J Med 318 (25): 1633-7, 1988.
- DiGiovanna JJ: Retinoid chemoprevention in the high-risk patient. J Am Acad Dermatol 39 (2 Pt 3): S82-5, 1998.
- DiGiovanna J: Oral isotretinoin chemoprevention of skin cancer in xeroderma pigmentosum. J Eur Acad Derm Venereol 5 (Suppl 1): 27, 1995.
- Otley CC, Stasko T, Tope WD, et al.: Chemoprevention of nonmelanoma skin cancer with systemic retinoids: practical dosing and management of adverse effects. Dermatol Surg 32 (4): 562-8, 2006.
- Mellerio JE, Robertson SJ, Bernardis C, et al.: Management of cutaneous squamous cell carcinoma in patients with epidermolysis bullosa: best clinical practice guidelines. Br J Dermatol 174 (1): 56-67, 2016.
- Yarosh D, Klein J, O'Connor A, et al.: Effect of topically applied T4 endonuclease V in liposomes on skin cancer in xeroderma pigmentosum: a randomised study. Xeroderma Pigmentosum Study Group. Lancet 357 (9260): 926-9, 2001.
- Boussen H, Zwik J, Mili-Boussen I, et al.: [Therapeutic results of 5-fluorouracil in multiple and unresectable facial carcinoma secondary to xeroderma pigmentosum] Therapie 56 (6): 751-4, 2001 Nov-Dec.
- Sarasin A: Progress and prospects of xeroderma pigmentosum therapy. Adv Exp Med Biol 637: 144-51, 2008.
- Paganelli A, Giordano E, Fiorentini C, et al.: Surgical management and oncological follow-up of cutaneous squamous cell carcinomas arising in epidermolysis bullosa patients. Int J Dermatol 61 (10): 1171-1174, 2022.
- Mellerio JE, Weiner M, Denyer JE, et al.: Medical management of epidermolysis bullosa: Proceedings of the IInd International Symposium on Epidermolysis Bullosa, Santiago, Chile, 2005. Int J Dermatol 46 (8): 795-800, 2007.
- Hammersen J, Neuner A, Wild F, et al.: Attenuation of Severe Generalized Junctional Epidermolysis Bullosa by Systemic Treatment with Gentamicin. Dermatology 235 (4): 315-322, 2019.
- Wagner JE, Ishida-Yamamoto A, McGrath JA, et al.: Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med 363 (7): 629-39, 2010.
- El-Darouti M, Fawzy M, Amin I, et al.: Treatment of dystrophic epidermolysis bullosa with bone marrow non-hematopoeitic stem cells: a randomized controlled trial. Dermatol Ther 29 (2): 96-100, 2016 Mar-Apr.
- Hainzl S, Peking P, Kocher T, et al.: COL7A1 Editing via CRISPR/Cas9 in Recessive Dystrophic Epidermolysis Bullosa. Mol Ther 25 (11): 2573-2584, 2017.
- Shinkuma S, Guo Z, Christiano AM: Site-specific genome editing for correction of induced pluripotent stem cells derived from dominant dystrophic epidermolysis bullosa. Proc Natl Acad Sci U S A 113 (20): 5676-81, 2016.
- Siprashvili Z, Nguyen NT, Gorell ES, et al.: Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients With Recessive Dystrophic Epidermolysis Bullosa. JAMA 316 (17): 1808-1817, 2016.
- Webber BR, Osborn MJ, McElroy AN, et al.: CRISPR/Cas9-based genetic correction for recessive dystrophic epidermolysis bullosa. NPJ Regen Med 1: , 2016.
- Mosallaei D, Hao M, Antaya RJ, et al.: Molecular and Clinical Outcomes After Intravenous Gentamicin Treatment for Patients With Junctional Epidermolysis Bullosa Caused by Nonsense Variants. JAMA Dermatol 158 (4): 366-374, 2022.
Melanoma
Introduction
Rare, high-penetrance and common, low-penetrance genetic factors for melanoma have been identified, and approximately 5% to 10% of all melanomas arise in multiple-case families. However, a significant fraction of these families do not have detectable pathogenic variants in specific susceptibility genes. The frequency with which multiple-case families are ascertained and specific genetic variants are identified differs substantially between populations and geographic regions. A major population-based study has concluded that the high-penetrance susceptibility geneCDKN2A does not make a large contribution to the incidence of melanoma.
Melanoma can also occur in other parts of the body, such as the eye and brain. For more information about melanoma of the eye, see Intraocular (Uveal) Melanoma Treatment.
Risk Factors for Melanoma
This section focuses on risk factors in individuals at increased hereditary risk of developing melanoma. For more information about melanoma risk factors in the general population, see Skin Cancer Prevention.
Sun exposure
Sun exposure is well established as a major etiologic factor in all forms of skin cancer, although its effects differ by cancer type. The relationship between sun exposure, sunscreen use, and the development of skin cancer is complex. It is complicated by negative confounding (i.e., subjects who are extremely sun sensitive deliberately engage in fewer activities in direct sunlight, and they are more likely to wear sunscreen when they do). These subjects are genetically susceptible to the development of skin cancer by virtue of their cutaneous phenotype and thus may develop skin cancer regardless of the amount of sunlight exposure or the sun protection factor of the sunscreen.
Pigmentary characteristics
Pigmentary characteristics are important determinants of melanoma susceptibility. There is an inverse correlation between melanoma risk and skin color that goes from lightest skin to darkest skin. Dark-skinned ethnic groups have a very low risk of melanoma on pigmented skin surfaces; however, individuals in these groups develop melanoma on less-pigmented acral surfaces (palms, soles, nail beds) at the same frequency as light-skinned individuals. Among relatively light-skinned individuals, skin color is modified by genetics and behavior. Melanocortin 1 receptor (MC1R) is one of the major genes controlling pigmentation. For more information, see the section on MC1R in the Melanoma section. Other pigmentation genes are under investigation. A relatively new area of investigation is the evaluation of multiple variants together, often referred to as a polygenic risk score (PRS). PRSs for cutaneous melanoma contain many genetic variants that impact the expression of genes important in pigment-related phenotypes. The integration of these models with known phenotype risk factors and their overall clinical utility in melanoma risk prediction have not been demonstrated. PRSs for melanoma are not clinically available at this time.
Clinically, several pigmentary characteristics are evaluated to assess the risk of melanoma and other types of skin cancer. These include the following:
- Fitzpatrick skin type. The following six skin phenotypes were defined on the basis of response to sun exposure at the beginning of summer.
- Type I: Extremely fair skin, always burns, never tans.
- Type II: Fair skin, always burns, sometimes tans.
- Type III: Medium skin, sometimes burns, always tans.
- Type IV: Olive skin, rarely burns, always tans.
- Type V: Moderately pigmented brown skin, never burns, always tans.
- Type VI: Markedly pigmented black skin, never burns, always tans.
- Number of nevi or nevus density.
- Abnormal or atypical nevi.
- Freckling.
Nevi
Nevi (or moles) are sharply circumscribed benign pigmented lesions of the skin or mucosa composed of nest melanocytes. Patients with multiple nevi demonstrate increased risk of melanoma. While there is evidence that both the presence of multiple nevi and the presence of multiple clinically atypical nevi are associated with an increased risk of melanoma, most studies demonstrate a stronger risk of melanoma with the presence of atypical nevi. In addition, patients with multiple atypical nevi, regardless of personal and/or family history of melanoma, are at significantly increased risk of developing melanoma than are patients without atypical nevi. A population-based study in the United Kingdom that identified genetic risk factors for the development of nevi showed that some of the same variants are modestly associated with melanoma risk.
The phenotype of multiple nevi has both familial and environmental affecters. The number of nevi can increase with childhood sun exposure. The analysis of this association is complex because the use of sun protection strongly correlates with sun exposure. Inheritance of the specific phenotype of a high number of nevi, including clinically atypical nevi, was initially reported as an autosomal dominant trait under the names dysplastic nevus syndrome and familial atypical multiple mole-melanoma syndrome. A portion of this inherited phenotype is attributed to the major melanoma risk gene CDKN2A discussed below. Even within gene carriers in high-risk families, sun exposure seems to affect nevus number.
Family history
Generally, a family history of melanoma appears to increase risk of melanoma by about twofold. A family cancer registry study assessed over 20,000 individuals with melanoma and found a standardized incidence ratio (SIR) of 2.62 for offspring of individuals with melanoma and 2.94 for siblings. A larger study of more than 200,000 individuals from the Nurses’ Health Study, the Nurses’ Health Study 2, and the Health Professionals Follow-up Study found that individuals with a family history of melanoma had an increased risk of melanoma (hazard ratio [HR], 1.74; 95% confidence interval [CI], 1.45–2.09). Slightly higher melanoma risks were found in a population-based study of 1,506,961 individuals in Western Australia; first-degree relatives (FDRs) of 5,660 individuals with melanoma showed an HR for melanoma of 3.58 (95% CI, 2.43–5.43). Another population-based study of more than 238,000 FDRs of 23,000 melanoma patients found a lifetime cumulative risk of melanoma of 2.5% to 3%, which is about double the risk of the general population. Risk based on family history is dependent not only on the number of individuals in the family who have a melanoma but also on the number of melanomas in each family member. For example, the familial risk of melanoma was found to increase 2.2-fold (95% CI, 2.2–2.3) with a single FDR who has one melanoma and up to 16.3-fold (95% CI, 9.5–26.1) with a single FDR who has five or more melanomas. When two or more family members were diagnosed with melanoma before age 30 years, the lifetime cumulative risk for the family members rose to 14%.
A study on the heritability of cancer among 80,309 monozygotic and 123,382 dizygotic twins showed that melanoma has a heritability of 58% (95% CI, 43%–73%), suggesting that more than half of the risk of melanoma is caused by inherited factors. A study looking at the contribution of family history to melanoma risk showed a population-attributable fraction ranging from less than 1% in northern Europe to 6.4% in Australia, suggesting that only a small percentage of melanoma cases are caused by familial factors. Rarely, however, in some families many generations and multiple individuals develop melanoma and are at much higher risk. For individuals from these families, the incidence of melanoma is higher for sun-protected rather than sun-exposed skin.
A major hereditary melanoma susceptibility gene, CDKN2A, is altered in approximately 14% to 43% of families with three or more melanoma cases. To date, pathogenic variants have not been identified in more than half of the families with multiple cases of melanoma. Multigene panel testing can be considered in families with multiple cases of melanoma. This type of testing can search for pathogenic variants in multiple melanoma genes. However, a large study of extended panel testing (30 genes) in melanoma families without CDKN2A and CDK4 pathogenic variants had a diagnostic yield of 4%. In a large Italian study of multigene panel testing in patients with melanoma, predictors of a pathogenic variant included the following: (1) the presence of pancreatic cancer and melanoma in the same family, and (2) individuals who have had more than three melanomas. When patients were diagnosed with melanoma after age 60 years, they were less likely to have a pathogenic variant in a melanoma gene.
The definition of a familial cluster of melanoma varies by geographical region worldwide, because of the role played by ultraviolet (UV) radiation in melanoma pathogenesis. In heavily insulated regions (regions with high ambient sun exposure), three or more affected family members are required for a familial cluster. In regions with lower levels of ambient sunlight, two or more affected family members are considered sufficient to define a familial cluster. For more information on reasons to consider genetic testing for melanoma, see the Genetic testing section.
Personal history of melanoma
A previous melanoma places one at high risk of developing additional primary melanomas, particularly for people with the most common risk factors for melanoma, such as cutaneous phenotype, family history, a pathogenic variant in CDKN2A, a great deal of early-life sun exposure, and numerous or atypical nevi. In the sporadic setting, approximately 5% of melanoma patients develop more than one primary cancer, while in the familial setting the corresponding estimate is 30%. This greater-than-expected rate of multiple primary cancers of the same organ is a common feature of hereditary cancer susceptibility syndromes; it represents a clinical finding that should raise the level of suspicion that a given patient’s melanoma may be related to an underlying genetic predisposition. Risk of a second primary melanoma after diagnosis of a first primary melanoma is approximately 5% and is greater for males and older patients. A study in Sweden of more than 65,000 individuals with melanoma found a SIR of 2.8 (95% CI, 2.3–3.4) for a second melanoma in individuals with a family history of melanoma and a SIR of 2.5 (95% CI, 2.3–2.7) in individuals with no family history. The risk of a second melanoma increased when the first melanoma was diagnosed before age 40 years (SIR, 4.7; 95% CI, 3.9–5.6%). The SIRs increased with increasing numbers of melanomas.
Personal history of BCC or SCC
Having a personal history of basal cell carcinoma (BCC) or squamous cell carcinoma (SCC) is also associated with an increase in risk of a subsequent melanoma. Depending on the study, this risk ranges from a nonsignificant increase for melanoma with a previous SCC of 1.04 (95% CI, 0.13–8.18) to a highly significant risk of 7.94 (95% CI, 4.11–15.35). It is likely that this relationship is the result of shared risk factors (of which sun exposure is presumably one), rather than a specific genetic factor that increases risk of both. Pigmentary characteristics are critically important for the development of melanoma, and cutaneous phenotype (described above), in combination with excessive sun exposure, is associated with an increased risk of all three types of skin cancers.
Major Genes for Melanoma
CDKN2A/p16 and p14/ARF
The major gene associated with melanoma is CDKN2A/p16, cyclin-dependent kinase inhibitor 2A, which is located on chromosome 9p21. This gene has multiple names (MTS1, INK4, and MLM) and is commonly called by the name of its protein, p16. It is an upstream regulator of the retinoblastoma gene pathway, acting through the cyclin D1/cyclin-dependent kinase 4 complex. This tumor suppressor gene has been intensively studied in multiple-case families and in population-based series of melanoma cases. CDKN2A controls the passage of cells through the cell cycle and provides a mechanism for holding damaged cells at the G1/S checkpoint to permit repair of DNA damage before cellular replication. Loss of function of tumor suppressor genes—a good example of which is CDKN2A—is a critical step in carcinogenesis for many tumor systems.
CDKN2A encodes two proteins, p16INK4a and p14ARF, both inhibitors of cellular senescence. The protein produced when the alternate reading frame (ARF) for exon 1 is transcribed instead of the standard reading frame exerts its biological effects through the p53 pathway. It mediates cell cycle arrest at the G1 and G2/M checkpoints, complementing p16’s block of G1/S progression—thereby facilitating cellular repair of DNA damage.
Pathogenic variants in CDKN2A account for 35% to 40% of familial melanomas and fewer than 1% of unselected melanoma cases. A study of more than 1,000 individuals in Spain showed that 6.6% of individuals with melanoma have a family history of two or more FDRs with melanoma, and up to 15% have a family history suggestive of familial melanoma that includes melanoma or pancreatic cancer diagnoses in FDRs or second-degree relatives (SDRs). A large case series from Britain found that CDKN2A pathogenic variants were present in 100% of families with seven to ten individuals affected with melanoma, 60% to 71% of families with four to six cases, and 14% of families with two cases. A similar study of Greek individuals with melanoma found CDKN2A pathogenic variants in 3.3% of single melanoma cases, 22% of familial melanoma cases, and 57% of individuals with multiple primary melanomas (MPM). A study of 92 sequential cases of Italian individuals with familial atypical multiple mole-melanoma syndrome (defined as three or more individuals with primary cutaneous melanoma or one individual with MPM) found CDKN2A pathogenic variants in 20% of individuals, including three unrelated individuals with a p.D84V variant. Cascade testing identified 14 of 40 unaffected family members undergoing testing who carried their family’s CDKN2A pathogenic variant. However, a second study of 106 familial melanoma cases (defined as at least two melanoma cases) only found CDKN2A pathogenic variants in 8.3% of cases. The frequency of CDKN2A pathogenic variants is as high as 22% in families with two cases of melanoma who have other features of hereditary melanoma, such as an age at diagnosis younger than 50 years or one or more individuals diagnosed with MPM. Many of the pathogenic variants reported among families are founder variants, which are unique to specific populations and geographic areas.
A study of 587 individuals with a single primary melanoma or MPM found CDKN2A pathogenic variants in 19% of individuals with MPM relative to 4.4% of individuals with a single primary melanoma. CDKN2A pathogenic variants were found in 29.6% of individuals with three or more primary melanomas. Fifty-eight percent of individuals with more than three primary melanomas and family histories of melanoma (undefined) had a CDKN2A pathogenic variant. A second, smaller study of 46 patients with three or more primary melanomas found CDKN2A pathogenic variants in 24% of participants.
Depending on the study design and target population, melanoma penetrance related to CDKN2A pathogenic variants differs widely. One study of 80 multiple-case families demonstrated that the penetrance varied by country, an observation that was attributed to major differences in sun exposure. For example, in Australia, the penetrance was 30% by age 50 years and 91% by age 80 years; in the United States, the penetrance was 50% by age 50 years and 76% by age 80 years; in Europe, the penetrance was 13% by age 50 years and 58% by age 80 years. In contrast, a comparison of families with the CDKN2A pathogenic variant in the United Kingdom and Australia demonstrated the same cumulative risk of melanoma; for CDKN2A carriers, the risk of developing melanoma seemed independent of ambient UV radiation. Another study of individuals with melanoma identified in eight population-based cancer registries and one hospital-based sample obtained a self-reported family history and sequenced CDKN2A in all individuals. The penetrance was estimated as 14% by age 50 years and 28% by age 80 years. The explanation for these differences lies in the method of identifying the individuals tested, with penetrance estimates increasing with the number of affected family members. The method of family ascertainment in the latter study made it much less likely that “heavily loaded” melanoma families would be identified. Coinheritance of MC1R variants also increases CDKN2A penetrance; this genetic variant, described in further detail below, is therefore both a low-penetrance susceptibility gene and a modifier gene. For more information, see the section on MC1R in the Melanoma section. Other modifier loci have also been assessed in CDKN2A carriers; interleukin-9 (IL9) and GSTT1 were the only loci with effects that reached statistical significance, suggesting that other minor risk factors may interact with major risk loci.
One study reported a melanoma incidence rate of 9.9 per 1,000 person years among 354 FDRs and 2.1 per 1,000 person years among 391 SDRs of probands with a p16-Leiden (c.225-243del19) CDKN2A pathogenic variant (95% CIs of 7.4–13.3 and 1.2–3.8, respectively). These data indicate a melanoma rate that is much higher than that of the general population (12.9-fold increased incidence) for SDRs in untested relatives of carriers of CDKN2A pathogenic variants.
A study compared the clinical features of 7,695 individuals with melanoma (182 individuals had a CDKN2A pathogenic variant and 7,513 did not have a CDKN2A pathogenic variant). Results showed that individuals with a CDKN2A pathogenic variant were significantly younger when they were diagnosed with melanoma (mean age at diagnosis for individuals with a CDKN2A pathogenic variant vs. individuals without a CDKN2A pathogenic variant, 39.0 y vs. 54.3 y; P< .001). Individuals with a CDKN2A pathogenic variant also had an increased chance of developing a second melanoma after 5 years when compared with the control group (5-year cumulative incidence rate for CDKN2A carriers, 23%; 5-year cumulative incidence rate for individuals in the control group, 2.3%). A study of pediatric patients with melanoma (ages 9–19 y) in melanoma-prone families also reported a significant increase in melanoma prevalence (6-fold to 28-fold) relative to the general population. In this series, 7 of 21 patients (33%) with CDKN2A pathogenic variants were diagnosed with MPMs before age 20 years. An Italian study performed genotype-phenotype correlations in 100 families with familial melanoma to determine clinical features that are predictive of CDKN2A pathogenic variants. Probands with MPMs, at least one melanoma with a Breslow thickness greater than 0.4 mm, and more than three affected family members had a greater than 90% likelihood of having a CDKN2A pathogenic variant; probands with none of these features had less than a 1% likelihood of having a CDKN2A pathogenic variant. Overall, the presence of MPMs was the most predictive feature in this study.
Carriers of CDKN2A pathogenic variants have melanomas that resemble sporadic melanomas. A large study that compared melanoma pathology between CDKN2A carriers and individuals with sporadic melanoma found few significant differences, with a minor trend of increased pigmentation among pathogenic variant carriers. Another study of more than 670 carriers of CDKN2A pathogenic variants and 1,258 carriers of wild-type or benign CDKN2A variants found that participants with pathogenic variants were more likely to be diagnosed at an earlier age (median age, 38 y vs. 46 y) and have MPM (average number of melanomas, 2.3 vs. 1.4). A small study compared the overall survival (OS) rates of 106 carriers of CDKN2A pathogenic variants and 199 noncarriers who did not have family histories of melanoma. CDKN2A carriers were more likely to have MPMs. However, there was no significant difference in OS or disease-specific survival rates between carriers and noncarriers. However, two pathogenic variants in CDKN2A (p.Arg112dup, p.Pro48Leu) may be prognostic factors in patients with melanoma. After adjusting for age, sex, and tumor classification, carriers of these CDKN2A pathogenic variants had poorer melanoma-specific survival than did non-CDKN2A carriers (HR, 2.5; 95% CI, 1.49–2.21). An early study suggested that somatic NRAS mutations occurred at a higher rate in melanomas diagnosed in Swedish families who carry CDKN2A pathogenic variants when compared with those who had sporadic melanomas. However, subsequent studies found that the rates of common somatic mutations (BRAF, NRAS) were lower in the melanomas of CDKN2A carriers than in the sporadic melanomas found in the control group. Several patients with CDKN2A variants had melanomas with coexisting BRAF and NRAS mutations; this is an uncommon occurrence in sporadic melanomas. Data from a small series of patients suggests that melanoma patients with CDKN2A pathogenic variants may have improved response rates to immunotherapy when compared with noncarriers; however, further data are needed in this area.
CDKN2A exon 1ß pathogenic variants (p14ARF) have been identified in a small percentage of families negative for p16INK4a pathogenic variants. In a study of 94 Italian families with two or more cases of melanoma, 3.2% of families had variants in p14ARF. A patient with a balanced translocation between chromosomes 9 and 22 that disrupted p14ARF had melanoma, DNA repair deficiency, and features of DiGeorge syndrome, including deafness and malformed inner ears.
There are models that can predict whether an individual has a pathogenic variant in CDKN2A. However, in the era of widely available and inexpensive multigene (panel) testing, these models are not widely utilized clinically.
CDKN2A, cutaneous phenotypes, and cancers other than melanoma
In a Melanoma Genetics Consortium (GenoMEL) study of 1,641 family members of melanoma probands, family members with a CDKN2A pathogenic variant were more likely to have atypical nevi than were family members of CDKN2A noncarriers (odds ratio [OR], 1.65; 95% CI, 1.18–2.28). Another study of individuals in Sweden with MPM and two or more cases of melanoma in their first-, second-, or third-degree relatives found CDKN2A pathogenic variants in 43 of 100 cases. Familial MPM cases with CDKN2A variants, familial MPM cases wild-type for CDKN2A, and nonfamilial MPM cases all showed increased risks of future cutaneous SCCs compared with controls (relative risk [RR], 4.8; 95% CI, 1.5–15.1).
Results from the Genes, Environment, and Melanoma (GEM) study showed that FDRs of carriers of CDKN2A pathogenic variants with melanoma had an approximately 50% increased risk of cancers other than melanoma, compared with FDRs of other melanoma patients. Cancers with increased risk in this population included gastrointestinal cancers (RR, 2.4; 95% CI, 1.4–3.7), pancreatic cancers (RR, 7.4; 95% CI, 2.3–18.7), and Wilms tumor (RR, 40.4; 95% CI, 3.4–352.7). A Spanish study of the FDRs of 66 melanoma patients with known CDKN2A pathogenic variants also showed an increase in prevalence of other cancers, including pancreatic (prevalence ratio [PR], 2.97; 95% CI, 1.72–5.15), lung (PR, 3.04; 95% CI, 1.93–4.80), and breast cancers (PR, 2.19; 95% CI, 1.36–3.55). A large registry study from Sweden that included 27 families carrying the Arg112dup pathogenic variant in CDKN2A observed excess nonmelanoma cancers in both carriers (n = 120) and FDRs (n = 275). For carriers of CDKN2A pathogenic variants, increased risks relative to a control population were seen for pancreatic (RR, 43.8; 95% CI, 13.8–139), upper digestive (RR, 17.1; 95% CI, 6.3–46.5), respiratory (RR, 15.6; 95% CI, 5.4–46.0), and breast cancers (RR 3.0; 95% CI, 0.9–9.9), among others (all cancers: RR, 5.0; 95% CI, 3.7–7.3). The RRs in FDRs were 20.6 (95% CI, 11.6–36.7) for pancreatic cancers, 6.0 (95% CI, 2.8–13.1) for respiratory cancers, 3.3 (95% CI, 1.5–7.6) for upper digestive cancers, and 1.9 (95% CI, 0.9–4.0) for breast cancers, with a RR of all cancers of 2.1 (95% CI, 1.6–2.7). A lesser-increased cancer risk was seen among SDRs. They also observed a significant association between smoking and risk of pancreatic, respiratory, and upper digestive cancers, with an OR of 9.3 (95% CI, 1.9–44.7) for ever-smoking carriers compared with never-smoking carriers. One study of individuals with CDKN2A pathogenic variants that affected both p16 and p14ARF transcripts found an increased incidence of esophageal cancers and malignant peripheral nerve sheath tumors, although the reported incidence for both of these tumors was very small.
A few studies have identified individuals with sarcoma who have germline pathogenic variants in CDKN2A, but the number of cases is too small to determine the risk of sarcoma associated with this gene. One patient with features of Li-Fraumeni syndrome did not carry a TP53 pathogenic variant, but a deletion of CDKN2A and CDKN2B. A whole-exome sequencing study of a Li-Fraumeni–like family with three individuals with soft tissue sarcoma identified a shared pathogenic CDKN2A variant. An evaluation of 474 melanoma families with cases of sarcoma and 190 TP53 variant–negative Li-Fraumeni–like families found eight additional individuals with sarcoma and pathogenic CDKN2A variants.
Pancreatic cancer
A subset of families carrying a CDKN2A pathogenic variant also displays an increased risk of pancreatic cancer. The overall lifetime risk of pancreatic cancer in these families ranges from 11% to 17%. The RR has been reported as high as 47.8. Although at least 18 different variants in p16 have been identified in such families, specific pathogenic variants appear to have a particularly elevated risk of pancreatic cancer. Pathogenic variants affecting splice sites or Ankyrin repeats were found more commonly in families with both pancreatic cancer and melanoma than in those with melanoma alone. The p16 Leiden variant is a 19-base pairdeletion in CDKN2A exon 2 and is a founder pathogenic variant originating in the Netherlands. In one major Dutch study, 19 families with 86 members who had melanoma also had 19 members with pancreatic cancer in their families, a cumulative risk of 17% by age 75 years. In this study, the median age of pancreatic cancer onset was 58 years, similar to the median age at onset for sporadic pancreatic cancer. However, other reports indicate that the average age at diagnosis is 5.8 years earlier for these carriers of pathogenic variants than for those with sporadic pancreatic cancer. Geographic variation may play a role in determining pancreatic risk in these families carrying known pathogenic variants. In a multicontinent study of the features of germlineCDKN2A pathogenic variants, Australian families carrying these variants did not have an increased risk of pancreatic cancer. It was also reported that similar CDKN2A variants were involved in families with and without pancreatic cancer; therefore, there are additional factors involved in the development of melanoma and pancreatic cancer. Some families with CDKN2A pathogenic variants may have a pattern of site-specific pancreatic cancer only. Conversely, melanoma-prone families that do not have a CDKN2A pathogenic variant have not been shown to have an increased risk of pancreatic cancer.
In a review of 110 families with multiple cases of pancreatic cancer, 18 showed an association between pancreatic cancer and melanoma. Only 5 of the 18 families with cases of both pancreatic cancer and melanoma had individuals with multiple dysplastic nevi. These 18 families were assessed for pathogenic variants in CDKN2A; variants were identified in only 2 of the 18 families, neither of which had a dysplastic nevi phenotype.
Melanoma-astrocytoma syndrome
The melanoma-astrocytoma syndrome is another phenotype caused by pathogenic variants in CDKN2A. The possible existence of this disorder was first described in 1993. A study of 904 individuals with melanoma and their families found 15 families with 17 members who had both melanoma and multiple types of tumors of the nervous system. Another study found a family with multiple melanoma and neural cell tumors that appeared to be caused by loss of p14ARF function or to disruption of expression of p16. Plexiform neurofibromas have also been reported in individuals with deleteriousCDKN2A variants.
CDK4 and CDK6
Cyclin-dependent kinases have important roles in progression of cells from G1 to S phase. CDK4 and CDK6 partner with the cyclin–D associated kinases to accelerate the function of the cell cycle. Phosphorylation of the retinoblastoma (Rb) protein in G1 by cyclin-dependent kinases releases transcription factors, inducing gene expression and metabolic changes that precede DNA replication, thus allowing the cell to progress through the cell cycle. These genes are of conceptual significance because they are in the same signaling pathway as CDKN2A.
Germline CDK4 pathogenic variants are very rare, being found in only a handful of melanoma kindreds. All described families demonstrated a substitution of amino acid 24, suggesting this position as a variant hotspot within the CDK4 gene. Three Latvian families with melanoma have a R24H substitution arising on the same haplotype, which suggests that it could be a founder pathogenic variant in this population. A CDK4 pathogenic variant affects binding of p16 with its subsequent inhibition of CDK4 functionality. With constitutive activation of germline CDK4, CDK4 acts as a dominant oncogene. A small study showed that the melanoma cancer risk in 17 families with CDK4 pathogenic variants was similar to the risk seen in families with CDKN2A variants. In addition, the melanomas found in CDK4 families appear to have similar rates of somatic BRAF mutations to those found in sporadic populations, although because of the rare nature of CDK4 germline variants, the data are necessarily limited. Despite many similarities, one study of 54 melanomas from 15 CDK4 pathogenic variant carriers, 348 melanomas from 141 CDKN2A pathogenic variant carriers, and 157 melanomas from 104 noncarriers found that tumors in CDK4 carriers were far more likely to arise on nontruncal areas (87% versus 45% in noncarriers [P< .0001] and 62% in CDKN2A carriers). For more information, see the CDKN2A/p16 and p14/ARF section.
Despite its functional similarity to CDK4, germline variants in CDK6 have not been identified in any melanoma kindreds.
Telomere maintenance genes
Telomerase reverse transcriptase (TERT)
While somatic activating variants in TERT are seen often in multiple cancer types, including sporadic melanoma, germline pathogenic variants in TERT are found rarely in melanoma families. A pathogenic variant in the promoter region of a TERT subunit was found in a single, large German kindred with multiple melanomas and other cancers (ovarian, renal, bladder, and lung). This pathogenic variant had increased promoter activity in construct assays. A detailed analysis of family members from this kindred was performed in 2022, and it emphasized the young ages at which melanoma was diagnosed in this kindred (median age at diagnosis, 30 y).
The frequency of TERT promoter variants in melanoma families has not been fully investigated, but one study of 273 families with three or more cases of melanoma identified only one family (with 7 melanoma cases) that carried a c.-57 T>G TERT promoter variant. Another study observed 202 Spanish families with two or more melanomas in which a TERT germline pathogenic variant was not identified.
The pathogenicity of rs2853669, a specific TERT variant, is up for interpretation since it has a high prevalence in the general population. The prevalence of this variant in the general population is estimated to be between 25% and 29%. A study of 106 familial melanoma cases (defined as at least two melanoma cases or MPM in the proband) found that 47% of MPM cases and 58% of familial melanoma cases carried this TERT promoter variant.
POT1
Exome and genome-sequencing in individuals from hereditary melanoma families led to the identification of missense pathogenic variants in POT1 that segregate with disease in numerous studies. A POT1 Ser270Asn missense pathogenic variant was found in 5 of 56 unrelated melanoma families from Italy. This variant was not observed in over 2,000 Italian controls. Ser270Asn is thought to be a founder pathogenic variant, as all families with the variant shared a haplotype. Additional POT1 missense pathogenic variants, including Tyr89Cys, Arg137His, and Gln623His, were identified in other melanoma families and were not seen in unaffected controls.
A study of 290 familial melanoma cases (defined as families with at least two melanomas) from the United States, Italy, and Spain identified 16 POT1 carriers in 10 families. Melanomas in POT1 carriers were more likely to have spitzoid morphology (P< .001) when compared with melanomas in the CDKN2A, CDK4, and non-carrier cohorts. This occurred even though melanomas were diagnosed at similar ages in the CDK4, CDKN2A, and POT1 cohorts. Additionally, melanomas in POT1 carriers were more likely to have moderate- to high-tumor–infiltrating lymphocytes (P< .001). POT1 pathogenic variants were found in approximately 1.7% to 13.5% of melanoma families who lacked CDKN2A or CDK4 pathogenic variants; therefore, POT1 may also be associated with hereditary melanoma. POT1 binds to single-stranded telomeric repeat regions and is thought to aid in maintenance of telomere length. Most of the variants segregating in families occur in the two oligonucleotide/oligosaccharide-binding domains of the protein, which are the portion of the protein critical for binding DNA. Individuals carrying POT1 pathogenic variants showed longer telomere lengths than melanoma cases without the POT1 variants, suggesting a link between disruption in normal telomere length and melanoma. The clinical utility of testing this gene has not yet been established.
ACD and TERF2IP
In one study, 510 melanoma families were screened by next-generation sequencing for pathogenic variants in genes in the shelterin complex, which protects chromosomal ends. Six families were found to have variants in ACD, and four families had variants in TERF2IP. The ACD variants clustered in the POT1 binding domain. Because some of these variants did not lead to a truncated protein, the functional significance is not confirmed.
DNA repair genes
Xeroderma pigmentosum (XP) patients with defective DNA repair have a more than 1,000-fold increase in melanoma risk. These patients are diagnosed with melanoma at a significantly younger age than individuals in the general population; on average, melanoma diagnosis occurs at age 22 years in XP patients. The anatomic site distribution of melanomas in XP patients is similar to that of the general population.
Genetic polymorphisms associated with DNA repair genes have been associated with mildly increased melanoma risk in the general population. A meta-analysis of eight case-control studies comprising more than 5,000 cases and 7,000 controls found that individuals carrying the Asp1104His polymorphism in XPG had an increased risk of melanoma (OR, 2.42; 95% CI, 2.26–2.60). For more information, see the section on Xeroderma pigmentosum in the Squamous Cell Carcinoma section.
BRCA1-associated protein 1 (BAP1)
BAP1 has recently emerged as a gene implicated both in sporadic and hereditary melanomas. Originally described in a cohort of uveal melanoma patients, BAP1 is a tumor suppressor gene that was found to be inactivated in 84% of uveal melanoma patients with metastases. Although the majority of these variants were somatic, one patient was found to have a germline frameshift variant. A phenotype associated with BAP1 pathogenic variants was subsequently described. Two families with multiple, elevated melanocytic tumors that were clinically and histopathologically distinct from other melanocytic neoplasms were found to have inactivating germline pathogenic variants of BAP1. These tumors, which have been termed melanocytic BAP1-mutated atypical intradermal tumors, or MBAITs, are found throughout the body, generally measure approximately 5 mm, and begin to appear in the second decade of life. MBAITs are 2 mm to 10 mm in diameter, and affected individuals (about 67% of BAP1 pathogenic variant carriers) can have 5 to more than 50 skin lesions. Cases of cutaneous melanoma were present in these families, but the rate of malignant progression is thought to be low due to the relative lack of melanomas in comparison with the number of more papular tumors. This syndrome has been called BAP1 tumor syndrome or the COMMON (cutaneous and ocular melanoma and atypical melanocytic proliferation with other internal neoplasms) syndrome, and it is inherited in an autosomal dominant pattern. Further investigation has supported the association between familial cutaneous melanoma and uveal melanoma in BAP1 carriers. However, potentially pathogenic BAP1 germline variants occur in a low percentage of melanoma cases. One targeted sequencing study of 1,109 unselected cutaneous melanoma cases found only seven germline missense pathogenic variants (<1%). A second series of 1,977 melanoma cases and 754 controls identified 22 rare variants in BAP1 among cases and 5 rare variants among controls; three of the variants found only among cases were confirmed to disrupt BAP1 function and were associated with family histories of other BAP1-associated cancers. In support of a link between melanoma risk and BAP1, in one series, about 18% of individuals with a BAP1 pathogenic variant developed melanoma. In addition, although data are currently limited, patients with germline pathogenic variants in BAP1 may be at increased risk of lung adenocarcinoma, mesothelioma, BCC, and clear cell carcinoma of the kidney.
Other studies have reported pathogenic variants in BAP1. A missense BAP1 pathogenic variant (p.Leu570Val) was described in a family with multiple cases of melanoma. This missense variant affected splicing and resulted in a truncated protein. This family also reported cases of uveal melanoma and paraganglioma. Another family with a Y646X BAP1 pathogenic variant reported multiple cancers including multiple cutaneous melanomas, BCCs, uveal melanomas, and mesotheliomas. The authors hypothesized that a genetic-environmental interaction between BAP1 pathogenic variants, UV radiation, and asbestos exposure contributed to the high incidence of cancers in this family.
A Markov simulation study estimated the survival and economic impacts of screening BAP1 pathogenic variant carriers for the four most common cancers associated with BAP1–predisposition syndrome (renal cancer, uveal melanoma, cutaneous melanoma, and mesothelioma). The researchers found that surveillance was cost-effective, costing approximately $1,265 per life-year gained. Survival also improved with BAP1-associated surveillance (50.2% of BAP1-related deaths occurred in the group that did not receive surveillance, whereas only 35.4% of BAP1-related deaths occurred in the group that received surveillance). The study estimated that only 171 deaths out of 10,000 individuals in the screened group were due to cutaneous melanoma, whereas 768 deaths out of 10,000 individuals in the nonscreened group were due to cutaneous melanoma.
Additional candidate regions for familial melanoma susceptibility
Several additional loci for familial melanoma have been identified through genome-wide studies. A melanoma susceptibility locus on 1p22 was identified through a linkage analysis of 49 Australian families who had at least three melanoma cases and who were negative for CDKN2A and CDK4 pathogenic variants. Deletion mapping in tumors shows a minimal region of loss of a 9-Mb interval within the peak linkage region, but none of the linkage families have pathogenic variants in the genes tested thus far. A genome-wide association study (GWAS) of individuals from 34 high-risk melanoma families revealed three single nucleotide variants (SNVs) on 10q25.1 associated with melanoma risk. The ORs for risk for the SNVs ranged from 6.8 to 8.4. Subsequent parametric linkage analysis in one family showed logarithm of the odd scores of 1.5, whereas the other two families assessed did not show linkage. No obvious candidate gene was identified in the genomic region of interest. Two genome-wide linkage studies of 35 and 42 Swedish families identified evidence of linkage on chromosomal regions 3q29, 17p11-12, and 18q22. An Italian linkage study showed a suggestive linkage to the same 3q29 locus. No causative genes have been confirmed, but candidates map to all of the loci. None of these loci have been confirmed in independent studies.
Several GWAS have suggested that a risk locus for melanoma may exist on chromosome 20q11 (OR, 1.27). This is the location of the ASIP locus that encodes the agouti signaling protein, which controls hair color during the hair growth cycle in some mammals. It acts as an antagonist to MC1R. While ASIP variation has been associated with differences in human pigmentation, initial studies did not find an association between ASIP variants and melanoma. However, other studies have shown that the ASIP rs56238684 variant is associated with MPM risk (OR, 2.5; 95% CI, 1.7–3.3). Additionally, variants in an ASIP transcription factor, NCOA6 (which is also on chromosome 20), had a maximum OR of 1.82 for melanoma risk. However, no interactions were seen between NCOA6 variants, MC1R variants, and melanoma risk. The mechanism by which variants at 20q11 cause increased melanoma risk remains unclear.
Other risk loci have been reported on chromosomes 2, 5, 6, 7, 9, 10, 11, 15, 16, and 22. A GWAS of melanoma published in 2014 examined eight of the loci with a previous significant association with melanoma, but without a confirmed causal gene. Researchers were able to confirm seven of eight loci and found some evidence supporting the eighth. These included the chromosome 20 locus discussed above and a 9p21 locus distinct from CDKN2A. Candidate genes at these loci seem to be clustered in functional groups associated with skin pigmentation and nevus development, both traits with a known melanoma association. For more information about these traits, see the Risk Factors for Melanoma section. A multicenter meta-analysis of 11 GWAS and two data sets included 15,990 cutaneous melanoma cases and 26,409 controls. They reported five melanoma susceptibility loci that involved putative melanocyte regulatory elements, telomere biology, and DNA repair.
A publicly available database, MelGene, maintains lists of variants that have been associated with melanoma risk through GWAS. MelGene also includes network and potential functional relationships between these genes and variants.
9q21 and GOLM1
When the first data linking CDKN2A pathogenic variants to melanoma risk became available, it was clear that these variants did not account for all the melanoma tumors in which 9p21 loss of heterozygosity could be demonstrated. In fact, 51% of informative cases had deletions that did not involve somatic mutations in CDKN2A. There are data that the golgi membrane protein 1 (GOLM1) gene, mapping to 9q21, may be involved in melanoma risk. Exome sequencing of DNA from 12 sets of cousins with cutaneous melanoma who were negative for known high-risk melanoma genes led to the identification of a rare GOLM1 variant (rs149739829) in three affected individuals in one pedigree. Two additional pairs of related melanoma cases with the putative risk allele were identified. Family-based case-control studies showed association with melanoma risk (OR, 9.81; P< .001). In a population-based case-control study of 1,534 melanoma cases, unselected for family history, and 1,146 controls, there was an increased risk of melanoma in individuals that carried the GOLM1 rs149739829 risk allele (OR, 2.45; P = .02).
Minor Genes (Genetic Modifiers) for Melanoma
MC1R
The MC1R gene, otherwise known as the alpha melanocyte-stimulating hormone receptor, is located on chromosome 8. Partial loss-of-function pathogenic variants, of which there are at least ten, are associated not only with red hair, fair skin, and poor tanning, but also with increased skin cancer risk independent of cutaneous pigmentation. A comprehensive meta-analysis of more than 8,000 cases and 50,000 controls showed the highest risk of melanoma in individuals with MC1R variants associated with red hair; however, alleles not associated with red hair have also been linked to increased melanoma risk. Additional phenotypic associations have been found. In different studies, MC1R variants were found to be associated with lentigo maligna melanoma (OR, 2.16; 95% CI, 1.07–4.37; P = .044) and increased risk of melanoma for individuals with no red hair, no freckles, and Fitzpatrick type III or IV skin (summary OR, 3.14; 95% CI, 2.06–4.80). Pooled studies of 5,160 cases and 12,119 controls from 17 sites calculated that melanoma risk attributable to MC1R variants is 28%, suggesting that these variants may be an important contributor to melanoma risk in the general population. In addition, individuals with MPMs have a high likelihood of carrying an MC1R pathogenic variant. In one study of 46 individuals with 3 or more primary melanomas, 43 individuals (93%) had an MC1R pathogenic variant.
A scoring system has been proposed for MC1R polymorphisms to identify associations between the degree of functional impairments in the melanogenesis pathway, clinical characteristics of patients, and melanoma presentations. The initial classification system designated MC1R variants that were strongly associated with red hair and fair skin as strong (R) variants (OR, 63.3; 95% CI, 31.9–139.6) and MC1R variants that were weakly associated with red hair and fair skin as weak (r) variants (OR, 5.1; 95% CI, 2.5–11.3). This work was expanded to evaluate additional MC1R variants, which were scored as follows: a consensus sequence allele was given a value of zero, an r allele was given a value of one, and an R allele was given a value of two. The summary score could range from zero to four. A study of 1,044 melanoma patients showed that individuals with a score of three or more were more likely to develop melanoma before age 50 years (OR, 1.47; 95% CI, 1.01–2.14). The MC1R score has been subsequently found to have implications for a survival benefit in melanoma patients. At least three studies have found a lower risk of death in melanoma patients without consensus MC1R alleles (HR, 0.78; 95% CI, 0.65–0.94) when compared with those with at least one MC1R consensus allele (HR, 0.57–0.78). However, the survival benefits associated with MC1R R alleles may only pertain to women.
MC1R variants are associated with an increased risk of all three types of skin cancer. However, adding MC1R genotype information to skin cancer risk predictions (which are based on age, sex, and cutaneous melanin density), only slightly improved them. In contrast, one study that examined predictors of early-onset melanoma in both population- and family-based studies showed that adding MC1R genotypes improved the area under the receiver operator curve (AUC) by 6% when compared with demographic information alone (P< .001). When MC1R genotypes were combined with an individual's nevi count and history of keratinocyte carcinoma (BCC or SCC), the AUC was 0.78 (95% CI, 0.75–0.82) for self-reported nevi and 0.83 (95% CI, 0.80–0.86) for physician-described nevi.
Several studies have tried to quantify the increased melanoma incidence seen in individuals with MC1R variants. An Australian study of 1,267 individuals found that those with an MC1R R/R genotype and greater than 20 nevi had a 25-fold increased risk of melanoma when compared with those who had a wildtype MC1R variant and had 0 to 4 nevi. Absolute melanoma risk (to age 75 years) was 23.3% in men with MC1R R/R genotypes and more than 20 nevi. Similarly, absolute melanoma risk (to age 75 years) was 19.3% in women with MC1R R/R genotypes and more than 20 nevi. A study of 233 young individuals with melanoma, 932 adults with melanoma, and 932 controls found that MC1R r variants were more common in young melanoma cases (≤20 years) when compared with adult patients aged 35 years and older (OR, 1.54; 95% CI, 1.02–2.33). The association was more pronounced in individuals aged 18 years and younger (OR, 1.80; 95% CI, 1.06–3.07).
A study of 1,791 individuals assessed if MC1R variants affect melanoma risk differently in men and women. Carrying two or more MC1R variants was associated with an increased risk in both women (OR, 2.65; 95% CI, 1.86–3.79; P = .001) and men (OR, 1.65; 95% CI, 1.14–2.38; P = .007). In a multivariate analysis (which included other risk factors like freckling, wrinkling, sunburns, and solar lentigines), the association between melanoma risk and MC1R variants was significant in women but not in men. A study in 203 individuals without skin cancer found that the MC1R r variant (p.Val60Leu) was associated with a high nevus count in women (>50 nevi; P< .001) but not in men. However, the number of individuals in the high-risk group was small.
MC1R variants can also modify melanoma risk in individuals with CDKN2A pathogenic variants. A study consisting of 815 carriers of CDKN2A pathogenic variants looked at four common non-synonymous MC1R variants and found that having one variant increased the melanoma risk twofold, but having two or more variants increased melanoma risk nearly sixfold. After stratification for hair color, the increased risk of melanoma appeared to be limited to subjects with brown or black hair. These data suggest that MC1R variants increase melanoma risk in a manner independent of their effect on pigmentation. A meta-analysis of individuals with CDKN2A pathogenic variants showed that those with greater than one variant in MC1R had approximately fourfold increased risk of melanoma. Individuals with one or more variants in MC1R showed an average 10-year decrease in age of onset from 47 to 37 years. In contrast, a large consortium study did not show as large a decrease in age at onset of melanoma. Another study of Norwegian melanoma cases and controls showed that carriers of CDKN2A pathogenic variants had an increased risk of melanoma when they carried either the Arg160Trp or Asp84Glu MC1R variants (R alleles).
In addition to studies evaluating the relationship between germline variants and MC1R variants, multiple groups have assessed whether MC1R variants are associated with somatic BRAF mutations. Studies indicate that there may be an association between MC1R variants and BRAF V600E somatic mutations. However, it is difficult to determine the impact of pigmentary influences on BRAF somatic mutations versus genetic effects.
Other pigmentary genes
Pathogenic variants in albinism genes may also account for a small proportion of familial melanoma. For example, variants in TYRP1, TYR, and OCA2 were observed at an increased frequency in one study of individuals with familial cutaneous melanoma compared with population controls. Further studies are needed to confirm these findings. For more information about pigmentary genes that can increase melanoma risk, see the Oculocutaneous albinism section.
MITF
Whole-genome sequencing led to the identification of the E318K variant in the microphthalmia–associated transcription factor (MITF) gene in a family with seven cases of melanoma. MITF is a transcription factor that regulates multiple genes that are pertinent to melanocyte function. The MITF E318K variant impairs normal SUMOylation of MITF. Individuals with the MITF E318K variant are more likely to have fair skin, high nevus counts, high freckling scores, and dark hair. Individuals with the MITF E318K variant are also more likely to develop clinically amelanotic melanomas, melanomas with nodular subtypes, and multiple primary melanomas. The MITF E318K variant is considered a moderate-risk melanoma allele with an OR ranging from 1.7 to 3.0 in cohorts from Australia, the United Kingdom, and Italy. However, MITF’s association with increased melanoma risk remains controversial, since not all studies have reproduced similar results. For example, a study in a Polish population of 4,266 cancer patients and 2,114 controls did not find an association between MITF pathogenic variants and melanoma.
Other Cancer Susceptibility Genes
BRCA1 and BRCA2
The Breast Cancer Linkage Consortium found that pathogenic variants in BRCA2 were associated with increased melanoma risk (RR, 2.58; 95% CI, 1.3–5.2). A second study reported that BRCA2 pathogenic variant carriers had a similar increase in melanoma risk, although the result fell short of statistical significance. In contrast, a large cohort study in the Netherlands showed that BRCA2 pathogenic variant carriers did not have increased melanoma risk; however, the expected incidence of melanoma was rare in this population, and this result reflects a difference of only two melanoma cases. Ashkenazi Jewish patients with melanoma do not have increased prevalence of the three common pathogenic founder variants in BRCA1 and BRCA2. It is unclear if BRCA1 and BRCA2 pathogenic variants increase melanoma risk based on the current evidence. For more information about the BRCA1 and BRCA2 genes, see BRCA1 and BRCA2: Cancer Risks and Management.
CHEK2
Studies have evaluated if other cancer susceptibility genes can increase an individual's risk of developing melanoma. A review of six mostly small retrospective studies of CHEK2, a gene that moderately increases risk for breast cancer and other cancers, did not show a consistent association with increased melanoma risk. The largest of these studies evaluated the CHEK2 c.1100delC pathogenic variant in Danish and German patients. This study included 1,905 melanoma cases and 12,860 controls. Data showed that individuals with the CHEK2 c.1100delC pathogenic variant had an increased risk of developing melanoma (OR, 1.7; 95% CI, 1.02–3.17). The researchers followed up with a meta-analysis of 2,619 cases and 17,481 controls. Similarly, CHEK2 c.1100delC pathogenic variant carriers had an increased risk of developing melanoma (OR, 1.81; 95% CI, 1.07–3.05). The other five studies did not find differences in CHEK2 pathogenic variant frequencies between cases and controls, had too few melanoma cases to make conclusions, and/or did not evaluate the CHEK2 c.1100delC pathogenic variant separately. For more information, see the CHEK2 section in Genetics of Breast and Gynecologic Cancers and the CHEK2 section in Genetics of Colorectal Cancer.
TP53 (Li-Fraumeni syndrome)
A longitudinal cohort study of 483 individuals with Li-Fraumeni syndrome identified 113 cutaneous malignancies including 42 BCCs, 26 SCCs, 21 melanomas, and 13 cutaneous sarcomas. In this cohort, the cumulative incidence of any skin cancer diagnosis was 36.3% by age 70 years. This corresponds to a 7.28-fold increased incidence of melanoma in individuals with Li-Fraumeni syndrome when compared with individuals in the general population. The median age of a melanoma diagnosis was age 42 years in this study (range, 21 y to 68 y), and for 7 of the 17 individuals with melanoma, it was their first cancer. SCC and BCC incidences were similar to those seen in the general population. For more information, see the Li-Fraumeni Syndrome section in Genetics of Breast and Gynecologic Cancers.
PTEN hamartoma tumor syndromes (including Cowden syndrome)
Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome (BRRS) are part of a spectrum of conditions known collectively as PTEN hamartoma tumor syndromes (PHTS). The term PHTS refers to any patient with a PTEN pathogenic variant, irrespective of clinical presentation. PTEN functions as a dual-specificity phosphatase that removes phosphate groups from tyrosine, serine, and threonine. Operational criteria for the diagnosis of Cowden syndrome have been published and subsequently updated. These include major, minor, and pathognomonic criteria, which consist of certain mucocutaneous manifestations such as trichilemmoma and adult-onset dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease). An updated set of criteria based on a systematic literature review has been suggested and is currently used in the National Comprehensive Cancer Network (NCCN) guidelines. For more information about PTEN hamartoma tumor syndromes (including Cowden syndrome), see Genetics of Colorectal Cancer and Genetics of Breast and Gynecologic Cancers.
The risk of melanoma in PTEN carriers is controversial. In an International Cowden Consortium (ICC) study of 100 patients, four women and four men were diagnosed with melanoma when less than one case of melanoma was expected; results showed a SIR of 28.3 for women (95% CI, 7.6–35.4) and 39.4 for men (95% CI, 10.6–100.9) (P< .001). In this ICC study, an elevated SIR of 8.5 (95% CI, 4.1–15.6) was reported in 368 PTEN pathogenic variant carriers. In this cohort, the estimated lifetime risk of melanoma in PTEN pathogenic variant carriers was 6% (range, 1.6%–9.4%). Notably, a subsequent prospective study did not observe an elevated melanoma risk in the same population. In this study, only 1 of 180 carriers was diagnosed with melanoma.
Melanoma Risk Assessment
Patients with a personal history of melanoma or dysplastic nevi should be asked to provide information regarding a family history of melanoma and other cancers to detect the presence of familial melanoma. Age at diagnosis in family members and pathologic confirmation, if available, should also be sought. The presence of MPM in the same individual may also provide a clue to an underlying genetic susceptibility. Approximately 30% of affected individuals in hereditary melanoma kindreds have more than one primary melanoma, versus 4% of sporadic melanoma patients. Family histories should be updated regularly; an annual review is often recommended.
For individuals without a personal history of melanoma, several models have been suggested for prediction of melanoma risk. The models differ in performance with respect to sensitivity and specificity, including differences by sex in some models. Data from the Nurses' Health Study were used to create a model that included gender, age, family history of melanoma, number of severe sunburns, number of moles larger than 3 mm on the limbs, and hair color. The concordance statistic for this model was 0.62 (95% CI, 0.58–0.65). Another measure of baseline melanoma risk was derived from a case-control study of individuals with and without melanoma in the Philadelphia and San Francisco areas. This model focused on gender, history of blistering sunburn, color of the complexion, number and size of moles, presence of freckling, presence of solar damage to the skin, absence of a tan, age, and geographic area of the United States. Attributable risk with this model was 86% for men and 89% for women. This predictive tool, the Melanoma Risk Assessment Tool, is available online. However, this tool was developed using a cohort of primarily White individuals without a personal or family history of melanoma or NMSC. It is designed for use by health professionals, and patients are encouraged to discuss results with their physicians. Additional external validation is appropriate before this tool can be adopted for widespread clinical use. Professional organizations have published genetic counseling referral guidelines for individuals with a history of melanoma. For more information, see the Family history section.
Genetic testing
Clinical testing is available to identify germline pathogenic variants in genes associated with hereditary melanoma such as MC1R, BAP1, BRCA2, CDK4, CDKN2A, MITF, TERT, and POT1. Multiple testing laboratories in the United States and overseas offer sequence analysis of the entire coding regions of these genes as well as deletion and duplication analysis. Unless there is a known pathogenic variant in a family, panel testing of multiple genes, rather than single gene sequencing, is often done. NCCN suggests considering genetic counseling and testing for CDKN2A if an individual has a personal or family history of either of the following: 1) three or more invasive cutaneous melanomas or 2) a combination of invasive melanoma, pancreatic cancer, and/or astrocytoma. Multigene (panel) testing may also be warranted when an individual has a family history of melanoma, and other cancers, including the following: uveal melanoma, astrocytoma, mesothelioma, breast cancer, pancreatic cancer, and/or renal cancer. Experts suggest that genetic testing be performed after genetic counseling is provided by a qualified genetics professional who is knowledgeable about hereditary skin cancer syndromes.
Interventions
High-risk population
Management of members of melanoma-prone families
High-risk individuals, including first- and second-degree family members in melanoma-prone families, should be educated about sun safety and warning signs of melanoma. Regular examination of the skin by a health care provider experienced in the evaluation of pigmented lesions is also recommended. One guideline suggests initiation of examination at age 10 years and conducting exams on a semiannual basis until nevi are considered stable, followed by annual examinations. These individuals should also be taught skin self-examination techniques, to be performed on a monthly basis. Observation of lesions may be aided by techniques such as full-body photography and dermoscopy. A cost-utility analysis has demonstrated the benefits of screening in this high-risk population. Additionally, surveillance has been shown to detect melanomas at an earlier stage than melanomas diagnosed before the identification of a CDKN2A pathogenic variant.
Biopsies of skin lesions in the high-risk population should be performed using the same criteria as those used for lesions in the general population. Prophylactic removal of nevi without clinically worrisome characteristics is not recommended. The reasons for this are practical: many individuals in these families have a large number of nevi, and complete removal of them all is not feasible, since new atypical nevi continue to develop. In addition, individuals with increased susceptibility to melanoma may have cancer arise de novo, without a precursor lesion such as a nevus.
Standard recommendations for screening and management of patients with BAP1 germline pathogenic variants are not currently available, but one group of experts has recommended annual ocular examinations starting at age 16 years, full-body skin examinations starting at age 20 years, and consideration of annual renal ultrasound and/or abdominal magnetic resonance imaging every 2 years.
Level of evidence: 5
At present, chemoprevention of melanoma in high-risk individuals remains an area of active investigation; however, no medications are recommended for melanoma risk reduction at this time.
Level of evidence: 5
Pancreatic cancer screening in CDKN2A pathogenic variant carriers
Screening for pancreatic cancer remains an area of investigation and controversy for carriers of CDKN2A pathogenic variants. At present, no effective means of pancreatic cancer screening is available for the general population; however, serum and radiographic screening measures are under study in high-risk populations. One proposed protocol suggested starting pancreatic screening in high-risk families at age 50 years or 10 years before the youngest age at diagnosis of pancreatic cancer in the family, whichever came first. In this algorithm, asymptomatic patients would be screened annually with serum cancer antigen 19-9 and endoscopic ultrasound, whereas symptomatic patients or those with abnormal test results would undergo endoscopic retrograde cholangiopancreatography (ERCP) and/or spiral computed tomography (CT) scanning. A study evaluating use of endoscopic ultrasound and ERCP in high-risk families concluded that these procedures were cost-effective in this setting.
The disadvantages of screening include the limitations of available noninvasive testing methods and the risks associated with invasive screening procedures. ERCP is the gold standard for identifying early cancers and precancerous lesions in the pancreas. However, serious complications such as bleeding, pancreatitis, and intestinal perforation can occur with this procedure. Implementation of pancreatic screening in carriers of CDKN2A pathogenic variants is further complicated by the apparent lack of increased incidence of pancreatic cancer in many of these families.
Most experts suggest that pancreatic cancer screening should be considered for carriers of CDKN2A pathogenic variants only if there is a family history of pancreatic cancer and, even then, only in the context of a clinical trial.
Level of evidence: 5
General population
Prevention, screening, and treatment interventions for melanoma in the general population (i.e., those who do not have a genetic predisposition to developing melanoma) are addressed in the following PDQ summaries:
- Skin Cancer Prevention
- Skin Cancer Screening
- Melanoma Treatment
References
- Berwick M, Orlow I, Hummer AJ, et al.: The prevalence of CDKN2A germ-line mutations and relative risk for cutaneous malignant melanoma: an international population-based study. Cancer Epidemiol Biomarkers Prev 15 (8): 1520-5, 2006.
- Rosso S, Zanetti R, Martinez C, et al.: The multicentre south European study 'Helios'. II: Different sun exposure patterns in the aetiology of basal cell and squamous cell carcinomas of the skin. Br J Cancer 73 (11): 1447-54, 1996.
- Berwick M: Counterpoint: sunscreen use is a safe and effective approach to skin cancer prevention. Cancer Epidemiol Biomarkers Prev 16 (10): 1923-4, 2007.
- Scherer D, Kumar R: Genetics of pigmentation in skin cancer--a review. Mutat Res 705 (2): 141-53, 2010.
- Cust AE, Drummond M, Kanetsky PA, et al.: Assessing the Incremental Contribution of Common Genomic Variants to Melanoma Risk Prediction in Two Population-Based Studies. J Invest Dermatol 138 (12): 2617-2624, 2018.
- Miles JA, Orlow I, Kanetsky PA, et al.: Relationship of Chromosome Arm 10q Variants to Occurrence of Multiple Primary Melanoma in the Population-Based Genes, Environment, and Melanoma (GEM) Study. J Invest Dermatol 139 (6): 1410-1412, 2019.
- Gu F, Chen TH, Pfeiffer RM, et al.: Combining common genetic variants and non-genetic risk factors to predict risk of cutaneous melanoma. Hum Mol Genet 27 (23): 4145-4156, 2018.
- Landi MT, Bishop DT, MacGregor S, et al.: Genome-wide association meta-analyses combining multiple risk phenotypes provide insights into the genetic architecture of cutaneous melanoma susceptibility. Nat Genet 52 (5): 494-504, 2020.
- Sordillo JE, Kraft P, Wu AC, et al.: Quantifying the Polygenic Contribution to Cutaneous Squamous Cell Carcinoma Risk. J Invest Dermatol 138 (7): 1507-1510, 2018.
- Fitzpatrick TB: The validity and practicality of sun-reactive skin types I through VI. Arch Dermatol 124 (6): 869-71, 1988.
- Roush GC, Nordlund JJ, Forget B, et al.: Independence of dysplastic nevi from total nevi in determining risk for nonfamilial melanoma. Prev Med 17 (3): 273-9, 1988.
- Halpern AC, Guerry D, Elder DE, et al.: Dysplastic nevi as risk markers of sporadic (nonfamilial) melanoma. A case-control study. Arch Dermatol 127 (7): 995-9, 1991.
- Garbe C, Büttner P, Weiss J, et al.: Risk factors for developing cutaneous melanoma and criteria for identifying persons at risk: multicenter case-control study of the Central Malignant Melanoma Registry of the German Dermatological Society. J Invest Dermatol 102 (5): 695-9, 1994.
- Tucker MA, Halpern A, Holly EA, et al.: Clinically recognized dysplastic nevi. A central risk factor for cutaneous melanoma. JAMA 277 (18): 1439-44, 1997.
- Marghoob AA, Kopf AW, Rigel DS, et al.: Risk of cutaneous malignant melanoma in patients with 'classic' atypical-mole syndrome. A case-control study. Arch Dermatol 130 (8): 993-8, 1994.
- Newton-Bishop JA, Chang YM, Iles MM, et al.: Melanocytic nevi, nevus genes, and melanoma risk in a large case-control study in the United Kingdom. Cancer Epidemiol Biomarkers Prev 19 (8): 2043-54, 2010.
- Azizi E, Iscovich J, Pavlotsky F, et al.: Use of sunscreen is linked with elevated naevi counts in Israeli school children and adolescents. Melanoma Res 10 (5): 491-8, 2000.
- Oliveria SA, Satagopan JM, Geller AC, et al.: Study of Nevi in Children (SONIC): baseline findings and predictors of nevus count. Am J Epidemiol 169 (1): 41-53, 2009.
- Elder DE, Goldman LI, Goldman SC, et al.: Dysplastic nevus syndrome: a phenotypic association of sporadic cutaneous melanoma. Cancer 46 (8): 1787-94, 1980.
- Lynch HT, Frichot BC, Lynch JF: Familial atypical multiple mole-melanoma syndrome. J Med Genet 15 (5): 352-6, 1978.
- Cannon-Albright LA, Meyer LJ, Goldgar DE, et al.: Penetrance and expressivity of the chromosome 9p melanoma susceptibility locus (MLM). Cancer Res 54 (23): 6041-4, 1994.
- Brandt A, Sundquist J, Hemminki K: Risk of incident and fatal melanoma in individuals with a family history of incident or fatal melanoma or any cancer. Br J Dermatol 165 (2): 342-8, 2011.
- Wei EX, Li X, Nan H: Having a first-degree relative with melanoma increases lifetime risk of melanoma, squamous cell carcinoma, and basal cell carcinoma. J Am Acad Dermatol 81 (2): 489-499, 2019.
- Ward SV, Dowty JG, Webster RJ, et al.: The aggregation of early-onset melanoma in young Western Australian families. Cancer Epidemiol 39 (3): 346-52, 2015.
- Chen T, Hemminki K, Kharazmi E, et al.: Multiple primary (even in situ) melanomas in a patient pose significant risk to family members. Eur J Cancer 50 (15): 2659-67, 2014.
- Fallah M, Pukkala E, Sundquist K, et al.: Familial melanoma by histology and age: joint data from five Nordic countries. Eur J Cancer 50 (6): 1176-83, 2014.
- Mucci LA, Hjelmborg JB, Harris JR, et al.: Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 315 (1): 68-76, 2016.
- Olsen CM, Carroll HJ, Whiteman DC: Familial melanoma: a meta-analysis and estimates of attributable fraction. Cancer Epidemiol Biomarkers Prev 19 (1): 65-73, 2010.
- Hemminki K, Zhang H, Czene K: Incidence trends and familial risks in invasive and in situ cutaneous melanoma by sun-exposed body sites. Int J Cancer 104 (6): 764-71, 2003.
- Goldstein AM, Chan M, Harland M, et al.: High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res 66 (20): 9818-28, 2006.
- Bishop JN, Harland M, Bishop DT: The genetics of melanoma. Br J Hosp Med (Lond) 67 (6): 299-304, 2006.
- Pissa M, Helkkula T, Appelqvist F, et al.: CDKN2A genetic testing in melanoma-prone families in Sweden in the years 2015-2020: implications for novel national recommendations. Acta Oncol 60 (7): 888-896, 2021.
- Bruno W, Dalmasso B, Barile M, et al.: Predictors of germline status for hereditary melanoma: 5 years of multi-gene panel testing within the Italian Melanoma Intergroup. ESMO Open 7 (4): 100525, 2022.
- Potjer TP, Bollen S, Grimbergen AJEM, et al.: Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer 144 (10): 2453-2464, 2019.
- Goggins WB, Tsao H: A population-based analysis of risk factors for a second primary cutaneous melanoma among melanoma survivors. Cancer 97 (3): 639-43, 2003.
- Slingluff CL, Vollmer RT, Seigler HF: Multiple primary melanoma: incidence and risk factors in 283 patients. Surgery 113 (3): 330-9, 1993.
- Giles G, Staples M, McCredie M, et al.: Multiple primary melanomas: an analysis of cancer registry data from Victoria and New South Wales. Melanoma Res 5 (6): 433-8, 1995.
- Begg CB, Orlow I, Hummer AJ, et al.: Lifetime risk of melanoma in CDKN2A mutation carriers in a population-based sample. J Natl Cancer Inst 97 (20): 1507-15, 2005.
- Chen T, Fallah M, Försti A, et al.: Risk of Next Melanoma in Patients With Familial and Sporadic Melanoma by Number of Previous Melanomas. JAMA Dermatol 151 (6): 607-15, 2015.
- Marghoob AA, Slade J, Salopek TG, et al.: Basal cell and squamous cell carcinomas are important risk factors for cutaneous malignant melanoma. Screening implications. Cancer 75 (2 Suppl): 707-14, 1995.
- Nugent Z, Demers AA, Wiseman MC, et al.: Risk of second primary cancer and death following a diagnosis of nonmelanoma skin cancer. Cancer Epidemiol Biomarkers Prev 14 (11 Pt 1): 2584-90, 2005.
- Rosenberg CA, Khandekar J, Greenland P, et al.: Cutaneous melanoma in postmenopausal women after nonmelanoma skin carcinoma: the Women's Health Initiative Observational Study. Cancer 106 (3): 654-63, 2006.
- Karagas MR, Greenberg ER, Mott LA, et al.: Occurrence of other cancers among patients with prior basal cell and squamous cell skin cancer. Cancer Epidemiol Biomarkers Prev 7 (2): 157-61, 1998.
- Chen J, Ruczinski I, Jorgensen TJ, et al.: Nonmelanoma skin cancer and risk for subsequent malignancy. J Natl Cancer Inst 100 (17): 1215-22, 2008.
- Aoude LG, Gartside M, Johansson P, et al.: Prevalence of Germline BAP1, CDKN2A, and CDK4 Mutations in an Australian Population-Based Sample of Cutaneous Melanoma Cases. Twin Res Hum Genet 18 (2): 126-33, 2015.
- Márquez-Rodas I, Martín González M, Nagore E, et al.: Frequency and characteristics of familial melanoma in Spain: the FAM-GEM-1 Study. PLoS One 10 (4): e0124239, 2015.
- Nikolaou V, Kang X, Stratigos A, et al.: Comprehensive mutational analysis of CDKN2A and CDK4 in Greek patients with cutaneous melanoma. Br J Dermatol 165 (6): 1219-22, 2011.
- Borroni RG, Manganoni AM, Grassi S, et al.: Genetic counselling and high-penetrance susceptibility gene analysis reveal the novel CDKN2A p.D84V (c.251A>T) mutation in melanoma-prone families from Italy. Melanoma Res 27 (2): 97-103, 2017.
- Pellegrini C, Maturo MG, Martorelli C, et al.: Characterization of melanoma susceptibility genes in high-risk patients from Central Italy. Melanoma Res 27 (3): 258-267, 2017.
- Maubec E, Chaudru V, Mohamdi H, et al.: Familial melanoma: clinical factors associated with germline CDKN2A mutations according to the number of patients affected by melanoma in a family. J Am Acad Dermatol 67 (6): 1257-64, 2012.
- Borg A, Johannsson U, Johannsson O, et al.: Novel germline p16 mutation in familial malignant melanoma in southern Sweden. Cancer Res 56 (11): 2497-500, 1996.
- Borg A, Sandberg T, Nilsson K, et al.: High frequency of multiple melanomas and breast and pancreas carcinomas in CDKN2A mutation-positive melanoma families. J Natl Cancer Inst 92 (15): 1260-6, 2000.
- Hashemi J, Bendahl PO, Sandberg T, et al.: Haplotype analysis and age estimation of the 113insR CDKN2A founder mutation in Swedish melanoma families. Genes Chromosomes Cancer 31 (2): 107-16, 2001.
- Ciotti P, Struewing JP, Mantelli M, et al.: A single genetic origin for the G101W CDKN2A mutation in 20 melanoma-prone families. Am J Hum Genet 67 (2): 311-9, 2000.
- Liu L, Dilworth D, Gao L, et al.: Mutation of the CDKN2A 5' UTR creates an aberrant initiation codon and predisposes to melanoma. Nat Genet 21 (1): 128-32, 1999.
- Pollock PM, Spurr N, Bishop T, et al.: Haplotype analysis of two recurrent CDKN2A mutations in 10 melanoma families: evidence for common founders and independent mutations. Hum Mutat 11 (6): 424-31, 1998.
- Larre Borges A, Borges AL, Cuéllar F, et al.: CDKN2A mutations in melanoma families from Uruguay. Br J Dermatol 161 (3): 536-41, 2009.
- de Ávila AL, Krepischi AC, Moredo LF, et al.: Germline CDKN2A mutations in Brazilian patients of hereditary cutaneous melanoma. Fam Cancer 13 (4): 645-9, 2014.
- Bruno W, Pastorino L, Ghiorzo P, et al.: Multiple primary melanomas (MPMs) and criteria for genetic assessment: MultiMEL, a multicenter study of the Italian Melanoma Intergroup. J Am Acad Dermatol 74 (2): 325-32, 2016.
- Li C, Liu T, Tavtigian SV, et al.: Targeted germline sequencing of patients with three or more primary melanomas reveals high rate of pathogenic variants. Melanoma Res 30 (3): 247-251, 2020.
- Bishop DT, Demenais F, Goldstein AM, et al.: Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst 94 (12): 894-903, 2002.
- Cust AE, Harland M, Makalic E, et al.: Melanoma risk for CDKN2A mutation carriers who are relatives of population-based case carriers in Australia and the UK. J Med Genet 48 (4): 266-72, 2011.
- Santillan AA, Cherpelis BS, Glass LF, et al.: Management of familial melanoma and nonmelanoma skin cancer syndromes. Surg Oncol Clin N Am 18 (1): 73-98, viii, 2009.
- Yang XR, Pfeiffer RM, Wheeler W, et al.: Identification of modifier genes for cutaneous malignant melanoma in melanoma-prone families with and without CDKN2A mutations. Int J Cancer 125 (12): 2912-7, 2009.
- Chaudru V, Lo MT, Lesueur F, et al.: Protective effect of copy number polymorphism of glutathione S-transferase T1 gene on melanoma risk in presence of CDKN2A mutations, MC1R variants and host-related phenotypes. Fam Cancer 8 (4): 371-7, 2009.
- van der Rhee JI, Boonk SE, Putter H, et al.: Surveillance of second-degree relatives from melanoma families with a CDKN2A germline mutation. Cancer Epidemiol Biomarkers Prev 22 (10): 1771-7, 2013.
- van der Rhee JI, Krijnen P, Gruis NA, et al.: Clinical and histologic characteristics of malignant melanoma in families with a germline mutation in CDKN2A. J Am Acad Dermatol 65 (2): 281-8, 2011.
- Goldstein AM, Stidd KC, Yang XR, et al.: Pediatric melanoma in melanoma-prone families. Cancer 124 (18): 3715-3723, 2018.
- Pedace L, De Simone P, Castori M, et al.: Clinical features predicting identification of CDKN2A mutations in Italian patients with familial cutaneous melanoma. Cancer Epidemiol 35 (6): e116-20, 2011.
- Sargen MR, Kanetsky PA, Newton-Bishop J, et al.: Histologic features of melanoma associated with CDKN2A genotype. J Am Acad Dermatol 72 (3): 496-507.e7, 2015.
- Taylor NJ, Handorf EA, Mitra N, et al.: Phenotypic and Histopathological Tumor Characteristics According to CDKN2A Mutation Status among Affected Members of Melanoma Families. J Invest Dermatol 136 (5): 1066-9, 2016.
- Dalmasso B, Pastorino L, Ciccarese G, et al.: CDKN2A germline mutations are not associated with poor survival in an Italian cohort of melanoma patients. J Am Acad Dermatol 80 (5): 1263-1271, 2019.
- Helgadottir H, Höiom V, Tuominen R, et al.: Germline CDKN2A Mutation Status and Survival in Familial Melanoma Cases. J Natl Cancer Inst 108 (11): , 2016.
- Eskandarpour M, Hashemi J, Kanter L, et al.: Frequency of UV-inducible NRAS mutations in melanomas of patients with germline CDKN2A mutations. J Natl Cancer Inst 95 (11): 790-8, 2003.
- Zebary A, Omholt K, van Doorn R, et al.: Somatic BRAF and NRAS mutations in familial melanomas with known germline CDKN2A status: a GenoMEL study. J Invest Dermatol 134 (1): 287-290, 2014.
- Jovanovic B, Egyhazi S, Eskandarpour M, et al.: Coexisting NRAS and BRAF mutations in primary familial melanomas with specific CDKN2A germline alterations. J Invest Dermatol 130 (2): 618-20, 2010.
- Helgadottir H, Ghiorzo P, van Doorn R, et al.: Efficacy of novel immunotherapy regimens in patients with metastatic melanoma with germline CDKN2A mutations. J Med Genet 57 (5): 316-321, 2020.
- Binni F, Antigoni I, De Simone P, et al.: Novel and recurrent p14 mutations in Italian familial melanoma. Clin Genet 77 (6): 581-6, 2010.
- Tan X, Anzick SL, Khan SG, et al.: Chimeric negative regulation of p14ARF and TBX1 by a t(9;22) translocation associated with melanoma, deafness, and DNA repair deficiency. Hum Mutat 34 (9): 1250-9, 2013.
- Niendorf KB, Goggins W, Yang G, et al.: MELPREDICT: a logistic regression model to estimate CDKN2A carrier probability. J Med Genet 43 (6): 501-6, 2006.
- Wang W, Niendorf KB, Patel D, et al.: Estimating CDKN2A carrier probability and personalizing cancer risk assessments in hereditary melanoma using MelaPRO. Cancer Res 70 (2): 552-9, 2010.
- Taylor NJ, Mitra N, Goldstein AM, et al.: Germline Variation at CDKN2A and Associations with Nevus Phenotypes among Members of Melanoma Families. J Invest Dermatol 137 (12): 2606-2612, 2017.
- Helgadottir H, Tuominen R, Olsson H, et al.: Cancer risks and survival in patients with multiple primary melanomas: Association with family history of melanoma and germline CDKN2A mutation status. J Am Acad Dermatol 77 (5): 893-901, 2017.
- Mukherjee B, Delancey JO, Raskin L, et al.: Risk of non-melanoma cancers in first-degree relatives of CDKN2A mutation carriers. J Natl Cancer Inst 104 (12): 953-6, 2012.
- Potrony M, Puig-Butillé JA, Aguilera P, et al.: Increased prevalence of lung, breast, and pancreatic cancers in addition to melanoma risk in families bearing the cyclin-dependent kinase inhibitor 2A mutation: implications for genetic counseling. J Am Acad Dermatol 71 (5): 888-95, 2014.
- Helgadottir H, Höiom V, Jönsson G, et al.: High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet 51 (8): 545-52, 2014.
- Sargen MR, Helgadottir H, Yang XR, et al.: Impact of Transcript (p16/p14ARF) Alteration on Cancer Risk in CDKN2A Germline Pathogenic Variant Carriers. JNCI Cancer Spectr 6 (6): , 2022.
- Jouenne F, Chauvot de Beauchene I, Bollaert E, et al.: Germline CDKN2A/P16INK4A mutations contribute to genetic determinism of sarcoma. J Med Genet 54 (9): 607-612, 2017.
- Chan SH, Lim WK, Michalski ST, et al.: Germline hemizygous deletion of CDKN2A-CDKN2B locus in a patient presenting with Li-Fraumeni syndrome. NPJ Genom Med 1: 16015, 2016.
- Goldstein AM, Fraser MC, Struewing JP, et al.: Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. N Engl J Med 333 (15): 970-4, 1995.
- Whelan AJ, Bartsch D, Goodfellow PJ: Brief report: a familial syndrome of pancreatic cancer and melanoma with a mutation in the CDKN2 tumor-suppressor gene. N Engl J Med 333 (15): 975-7, 1995.
- Lindor NM, McMaster ML, Lindor CJ, et al.: Concise handbook of familial cancer susceptibility syndromes - second edition. J Natl Cancer Inst Monogr (38): 1-93, 2008.
- de Snoo FA, Bishop DT, Bergman W, et al.: Increased risk of cancer other than melanoma in CDKN2A founder mutation (p16-Leiden)-positive melanoma families. Clin Cancer Res 14 (21): 7151-7, 2008.
- Goldstein AM: Familial melanoma, pancreatic cancer and germline CDKN2A mutations. Hum Mutat 23 (6): 630, 2004.
- Vasen HF, Gruis NA, Frants RR, et al.: Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int J Cancer 87 (6): 809-11, 2000.
- McWilliams RR, Bamlet WR, Rabe KG, et al.: Association of family history of specific cancers with a younger age of onset of pancreatic adenocarcinoma. Clin Gastroenterol Hepatol 4 (9): 1143-7, 2006.
- Goldstein AM, Chan M, Harland M, et al.: Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents. J Med Genet 44 (2): 99-106, 2007.
- Goldstein AM, Struewing JP, Chidambaram A, et al.: Genotype-phenotype relationships in U.S. melanoma-prone families with CDKN2A and CDK4 mutations. J Natl Cancer Inst 92 (12): 1006-10, 2000.
- Bartsch DK, Sina-Frey M, Lang S, et al.: CDKN2A germline mutations in familial pancreatic cancer. Ann Surg 236 (6): 730-7, 2002.
- Harinck F, Kluijt I, van der Stoep N, et al.: Indication for CDKN2A-mutation analysis in familial pancreatic cancer families without melanomas. J Med Genet 49 (6): 362-5, 2012.
- Zhen DB, Rabe KG, Gallinger S, et al.: BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genet Med 17 (7): 569-77, 2015.
- Bartsch DK, Langer P, Habbe N, et al.: Clinical and genetic analysis of 18 pancreatic carcinoma/melanoma-prone families. Clin Genet 77 (4): 333-41, 2010.
- Kaufman DK, Kimmel DW, Parisi JE, et al.: A familial syndrome with cutaneous malignant melanoma and cerebral astrocytoma. Neurology 43 (9): 1728-31, 1993.
- Azizi E, Friedman J, Pavlotsky F, et al.: Familial cutaneous malignant melanoma and tumors of the nervous system. A hereditary cancer syndrome. Cancer 76 (9): 1571-8, 1995.
- Randerson-Moor JA, Harland M, Williams S, et al.: A germline deletion of p14(ARF) but not CDKN2A in a melanoma-neural system tumour syndrome family. Hum Mol Genet 10 (1): 55-62, 2001.
- Vanneste R, Smith E, Graham G: Multiple neurofibromas as the presenting feature of familial atypical multiple malignant melanoma (FAMMM) syndrome. Am J Med Genet A 161A (6): 1425-31, 2013.
- Petty EM, Bolognia JL, Bale AE, et al.: Cutaneous malignant melanoma and atypical moles associated with a constitutional rearrangement of chromosomes 5 and 9. Am J Med Genet 45 (1): 77-80, 1993.
- Petronzelli F, Sollima D, Coppola G, et al.: CDKN2A germline splicing mutation affecting both p16(ink4) and p14(arf) RNA processing in a melanoma/neurofibroma kindred. Genes Chromosomes Cancer 31 (4): 398-401, 2001.
- Bahuau M, Vidaud D, Jenkins RB, et al.: Germ-line deletion involving the INK4 locus in familial proneness to melanoma and nervous system tumors. Cancer Res 58 (11): 2298-303, 1998.
- Zuo L, Weger J, Yang Q, et al.: Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet 12 (1): 97-9, 1996.
- Soufir N, Avril MF, Chompret A, et al.: Prevalence of p16 and CDK4 germline mutations in 48 melanoma-prone families in France. The French Familial Melanoma Study Group. Hum Mol Genet 7 (2): 209-16, 1998.
- Molven A, Grimstvedt MB, Steine SJ, et al.: A large Norwegian family with inherited malignant melanoma, multiple atypical nevi, and CDK4 mutation. Genes Chromosomes Cancer 44 (1): 10-8, 2005.
- Veinalde R, Ozola A, Azarjana K, et al.: Analysis of Latvian familial melanoma patients shows novel variants in the noncoding regions of CDKN2A and that the CDK4 mutation R24H is a founder mutation. Melanoma Res 23 (3): 221-6, 2013.
- Puntervoll HE, Yang XR, Vetti HH, et al.: Melanoma prone families with CDK4 germline mutation: phenotypic profile and associations with MC1R variants. J Med Genet 50 (4): 264-70, 2013.
- Puntervoll HE, Molven A, Akslen LA: Frequency of somatic BRAF mutations in melanocytic lesions from patients in a CDK4 melanoma family. Pigment Cell Melanoma Res 27 (1): 149-51, 2014.
- Sargen MR, Pfeiffer RM, Yang XR, et al.: Variation in Cutaneous Patterns of Melanomagenesis According to Germline CDKN2A/CDK4 Status in Melanoma-Prone Families. J Invest Dermatol 140 (1): 174-181.e3, 2020.
- Shennan MG, Badin AC, Walsh S, et al.: Lack of germline CDK6 mutations in familial melanoma. Oncogene 19 (14): 1849-52, 2000.
- Huang FW, Hodis E, Xu MJ, et al.: Highly recurrent TERT promoter mutations in human melanoma. Science 339 (6122): 957-9, 2013.
- Horn S, Figl A, Rachakonda PS, et al.: TERT promoter mutations in familial and sporadic melanoma. Science 339 (6122): 959-61, 2013.
- Zaremba A, Meier F, Schlein C, et al.: Clinical and pathological characteristics of familial melanoma with germline TERT promoter variants. Pigment Cell Melanoma Res 35 (6): 573-586, 2022.
- Harland M, Petljak M, Robles-Espinoza CD, et al.: Germline TERT promoter mutations are rare in familial melanoma. Fam Cancer 15 (1): 139-44, 2016.
- Potrony M, Puig-Butille JA, Ribera-Sola M, et al.: POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol 181 (1): 105-113, 2019.
- Reference SNP (refSNP) Cluster Report: rs2853669. In: Database of Short Genetic Variations (dbSNP) [Database]. Bethesda, MD: National Center for Biotechnology Information, 2018. Available online. Last accessed August 22, 2023.
- Shi J, Yang XR, Ballew B, et al.: Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet 46 (5): 482-6, 2014.
- Robles-Espinoza CD, Harland M, Ramsay AJ, et al.: POT1 loss-of-function variants predispose to familial melanoma. Nat Genet 46 (5): 478-81, 2014.
- Sargen MR, Calista D, Elder DE, et al.: Histologic features of melanoma associated with germline mutations of CDKN2A, CDK4, and POT1 in melanoma-prone families from the United States, Italy, and Spain. J Am Acad Dermatol 83 (3): 860-869, 2020.
- Aoude LG, Pritchard AL, Robles-Espinoza CD, et al.: Nonsense mutations in the shelterin complex genes ACD and TERF2IP in familial melanoma. J Natl Cancer Inst 107 (2): , 2015.
- Bradford PT, Goldstein AM, Tamura D, et al.: Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet 48 (3): 168-76, 2011.
- Kraemer KH, Lee MM, Scotto J: DNA repair protects against cutaneous and internal neoplasia: evidence from xeroderma pigmentosum. Carcinogenesis 5 (4): 511-4, 1984.
- Kraemer KH, Lee MM, Andrews AD, et al.: The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Dermatol 130 (8): 1018-21, 1994.
- Blankenburg S, König IR, Moessner R, et al.: Assessment of 3 xeroderma pigmentosum group C gene polymorphisms and risk of cutaneous melanoma: a case-control study. Carcinogenesis 26 (6): 1085-90, 2005.
- Xu Y, Jiao G, Wei L, et al.: Current evidences on the XPG Asp1104His polymorphism and melanoma susceptibility: a meta-analysis based on case-control studies. Mol Genet Genomics 290 (1): 273-9, 2015.
- Walpole S, Pritchard AL, Cebulla CM, et al.: Comprehensive Study of the Clinical Phenotype of Germline BAP1 Variant-Carrying Families Worldwide. J Natl Cancer Inst 110 (12): 1328-1341, 2018.
- Harbour JW, Onken MD, Roberson ED, et al.: Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 330 (6009): 1410-3, 2010.
- Wiesner T, Obenauf AC, Murali R, et al.: Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet 43 (10): 1018-21, 2011.
- Soura E, Eliades PJ, Shannon K, et al.: Hereditary melanoma: Update on syndromes and management: Emerging melanoma cancer complexes and genetic counseling. J Am Acad Dermatol 74 (3): 411-20; quiz 421-2, 2016.
- Njauw CN, Kim I, Piris A, et al.: Germline BAP1 inactivation is preferentially associated with metastatic ocular melanoma and cutaneous-ocular melanoma families. PLoS One 7 (4): e35295, 2012.
- Abdel-Rahman MH, Pilarski R, Cebulla CM, et al.: Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet 48 (12): 856-9, 2011.
- Carbone M, Ferris LK, Baumann F, et al.: BAP1 cancer syndrome: malignant mesothelioma, uveal and cutaneous melanoma, and MBAITs. J Transl Med 10: 179, 2012.
- Testa JR, Cheung M, Pei J, et al.: Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet 43 (10): 1022-5, 2011.
- Popova T, Hebert L, Jacquemin V, et al.: Germline BAP1 mutations predispose to renal cell carcinomas. Am J Hum Genet 92 (6): 974-80, 2013.
- Aoude LG, Wadt K, Bojesen A, et al.: A BAP1 mutation in a Danish family predisposes to uveal melanoma and other cancers. PLoS One 8 (8): e72144, 2013.
- O'Shea SJ, Robles-Espinoza CD, McLellan L, et al.: A population-based analysis of germline BAP1 mutations in melanoma. Hum Mol Genet 26 (4): 717-728, 2017.
- Wadt K, Choi J, Chung JY, et al.: A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanoma, and paraganglioma. Pigment Cell Melanoma Res 25 (6): 815-8, 2012.
- Rai K, Pilarski R, Cebulla CM, et al.: Comprehensive review of BAP1 tumor predisposition syndrome with report of two new cases. Clin Genet 89 (3): 285-94, 2016.
- Cheung M, Kadariya Y, Talarchek J, et al.: Germline BAP1 mutation in a family with high incidence of multiple primary cancers and a potential gene-environment interaction. Cancer Lett 369 (2): 261-5, 2015.
- Walpole S, Hayward NK, Pritchard AL, et al.: Microsimulation Model for Evaluating the Cost-Effectiveness of Surveillance in BAP1 Pathogenic Variant Carriers. JCO Clin Cancer Inform 5: 143-154, 2021.
- Gillanders E, Juo SH, Holland EA, et al.: Localization of a novel melanoma susceptibility locus to 1p22. Am J Hum Genet 73 (2): 301-13, 2003.
- Walker GJ, Indsto JO, Sood R, et al.: Deletion mapping suggests that the 1p22 melanoma susceptibility gene is a tumor suppressor localized to a 9-Mb interval. Genes Chromosomes Cancer 41 (1): 56-64, 2004.
- Teerlink C, Farnham J, Allen-Brady K, et al.: A unique genome-wide association analysis in extended Utah high-risk pedigrees identifies a novel melanoma risk variant on chromosome arm 10q. Hum Genet 131 (1): 77-85, 2012.
- Höiom V, Tuominen R, Hansson J: Genome-wide linkage analysis of Swedish families to identify putative susceptibility loci for cutaneous malignant melanoma. Genes Chromosomes Cancer 50 (12): 1076-84, 2011.
- Tuominen R, Jönsson G, Enerbäck C, et al.: Investigation of a putative melanoma susceptibility locus at chromosome 3q29. Cancer Genet 207 (3): 70-4, 2014.
- Potrony M, Puig-Butille JA, Farnham JM, et al.: Genome-wide linkage analysis in Spanish melanoma-prone families identifies a new familial melanoma susceptibility locus at 11q. Eur J Hum Genet 26 (8): 1188-1193, 2018.
- Gudbjartsson DF, Sulem P, Stacey SN, et al.: ASIP and TYR pigmentation variants associate with cutaneous melanoma and basal cell carcinoma. Nat Genet 40 (7): 886-91, 2008.
- Maccioni L, Rachakonda PS, Scherer D, et al.: Variants at chromosome 20 (ASIP locus) and melanoma risk. Int J Cancer 132 (1): 42-54, 2013.
- Kanetsky PA, Swoyer J, Panossian S, et al.: A polymorphism in the agouti signaling protein gene is associated with human pigmentation. Am J Hum Genet 70 (3): 770-5, 2002.
- Landi MT, Kanetsky PA, Tsang S, et al.: MC1R, ASIP, and DNA repair in sporadic and familial melanoma in a Mediterranean population. J Natl Cancer Inst 97 (13): 998-1007, 2005.
- McMeniman EK, Duffy DL, Jagirdar K, et al.: The interplay of sun damage and genetic risk in Australian multiple and single primary melanoma cases and controls. Br J Dermatol 183 (2): 357-366, 2020.
- Bishop DT, Demenais F, Iles MM, et al.: Genome-wide association study identifies three loci associated with melanoma risk. Nat Genet 41 (8): 920-5, 2009.
- Brown KM, Macgregor S, Montgomery GW, et al.: Common sequence variants on 20q11.22 confer melanoma susceptibility. Nat Genet 40 (7): 838-40, 2008.
- Falchi M, Bataille V, Hayward NK, et al.: Genome-wide association study identifies variants at 9p21 and 22q13 associated with development of cutaneous nevi. Nat Genet 41 (8): 915-9, 2009.
- Fernandez LP, Milne RL, Pita G, et al.: SLC45A2: a novel malignant melanoma-associated gene. Hum Mutat 29 (9): 1161-7, 2008.
- Kocarnik JM, Park SL, Han J, et al.: Replication of associations between GWAS SNPs and melanoma risk in the Population Architecture Using Genomics and Epidemiology (PAGE) Study. J Invest Dermatol 134 (7): 2049-52, 2014.
- Law MH, Bishop DT, Lee JE, et al.: Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat Genet 47 (9): 987-95, 2015.
- Gerstenblith MR, Shi J, Landi MT: Genome-wide association studies of pigmentation and skin cancer: a review and meta-analysis. Pigment Cell Melanoma Res 23 (5): 587-606, 2010.
- Athanasiadis EI, Antonopoulou K, Chatzinasiou F, et al.: A Web-based database of genetic association studies in cutaneous melanoma enhanced with network-driven data exploration tools. Database (Oxford) 2014: , 2014.
- Ohta M, Berd D, Shimizu M, et al.: Deletion mapping of chromosome region 9p21-p22 surrounding the CDKN2 locus in melanoma. Int J Cancer 65 (6): 762-7, 1996.
- Teerlink CC, Huff C, Stevens J, et al.: A Nonsynonymous Variant in the GOLM1 Gene in Cutaneous Malignant Melanoma. J Natl Cancer Inst 110 (12): 1380-1385, 2018.
- Box NF, Duffy DL, Chen W, et al.: MC1R genotype modifies risk of melanoma in families segregating CDKN2A mutations. Am J Hum Genet 69 (4): 765-73, 2001.
- Scherer D, Nagore E, Bermejo JL, et al.: Melanocortin receptor 1 variants and melanoma risk: a study of 2 European populations. Int J Cancer 125 (8): 1868-75, 2009.
- Kanetsky PA, Panossian S, Elder DE, et al.: Does MC1R genotype convey information about melanoma risk beyond risk phenotypes? Cancer 116 (10): 2416-28, 2010.
- Tagliabue E, Fargnoli MC, Gandini S, et al.: MC1R gene variants and non-melanoma skin cancer: a pooled-analysis from the M-SKIP project. Br J Cancer 113 (2): 354-63, 2015.
- Williams PF, Olsen CM, Hayward NK, et al.: Melanocortin 1 receptor and risk of cutaneous melanoma: a meta-analysis and estimates of population burden. Int J Cancer 129 (7): 1730-40, 2011.
- Puig-Butillé JA, Carrera C, Kumar R, et al.: Distribution of MC1R variants among melanoma subtypes: p.R163Q is associated with lentigo maligna melanoma in a Mediterranean population. Br J Dermatol 169 (4): 804-11, 2013.
- Pasquali E, García-Borrón JC, Fargnoli MC, et al.: MC1R variants increased the risk of sporadic cutaneous melanoma in darker-pigmented Caucasians: a pooled-analysis from the M-SKIP project. Int J Cancer 136 (3): 618-31, 2015.
- Duffy DL, Box NF, Chen W, et al.: Interactive effects of MC1R and OCA2 on melanoma risk phenotypes. Hum Mol Genet 13 (4): 447-61, 2004.
- Davies JR, Randerson-Moor J, Kukalizch K, et al.: Inherited variants in the MC1R gene and survival from cutaneous melanoma: a BioGenoMEL study. Pigment Cell Melanoma Res 25 (3): 384-94, 2012.
- Peña-Vilabelda MM, García-Casado Z, Requena C, et al.: Clinical characteristics of patients with cutaneous melanoma according to variants in the melanocortin 1 receptor gene. Actas Dermosifiliogr 105 (2): 159-71, 2014.
- Taylor NJ, Reiner AS, Begg CB, et al.: Inherited variation at MC1R and ASIP and association with melanoma-specific survival. Int J Cancer 136 (11): 2659-67, 2015.
- Lira FE, Podlipnik S, Potrony M, et al.: Inherited MC1R variants in patients with melanoma are associated with better survival in women. Br J Dermatol 182 (1): 138-146, 2020.
- Dwyer T, Stankovich JM, Blizzard L, et al.: Does the addition of information on genotype improve prediction of the risk of melanoma and nonmelanoma skin cancer beyond that obtained from skin phenotype? Am J Epidemiol 159 (9): 826-33, 2004.
- Han J, Kraft P, Colditz GA, et al.: Melanocortin 1 receptor variants and skin cancer risk. Int J Cancer 119 (8): 1976-84, 2006.
- Cust AE, Goumas C, Vuong K, et al.: MC1R genotype as a predictor of early-onset melanoma, compared with self-reported and physician-measured traditional risk factors: an Australian case-control-family study. BMC Cancer 13: 406, 2013.
- Duffy DL, Lee KJ, Jagirdar K, et al.: High naevus count and MC1R red hair alleles contribute synergistically to increased melanoma risk. Br J Dermatol 181 (5): 1009-1016, 2019.
- Pellegrini C, Botta F, Massi D, et al.: MC1R variants in childhood and adolescent melanoma: a retrospective pooled analysis of a multicentre cohort. Lancet Child Adolesc Health 3 (5): 332-342, 2019.
- Wendt J, Mueller C, Rauscher S, et al.: Contributions by MC1R Variants to Melanoma Risk in Males and Females. JAMA Dermatol 154 (7): 789-795, 2018.
- van der Poel LAJ, Bergman W, Gruis NA, et al.: The role of MC1R gene variants and phenotypical features in predicting high nevus count. Melanoma Res 30 (5): 511-514, 2020.
- Demenais F, Mohamdi H, Chaudru V, et al.: Association of MC1R variants and host phenotypes with melanoma risk in CDKN2A mutation carriers: a GenoMEL study. J Natl Cancer Inst 102 (20): 1568-83, 2010.
- Fargnoli MC, Gandini S, Peris K, et al.: MC1R variants increase melanoma risk in families with CDKN2A mutations: a meta-analysis. Eur J Cancer 46 (8): 1413-20, 2010.
- Helsing P, Nymoen DA, Rootwelt H, et al.: MC1R, ASIP, TYR, and TYRP1 gene variants in a population-based series of multiple primary melanomas. Genes Chromosomes Cancer 51 (7): 654-61, 2012.
- Fargnoli MC, Fargnoli MC, Pike K, et al.: MC1R variants increase risk of melanomas harboring BRAF mutations. J Invest Dermatol 128 (10): 2485-90, 2008.
- Guida M, Strippoli S, Ferretta A, et al.: Detrimental effects of melanocortin-1 receptor (MC1R) variants on the clinical outcomes of BRAF V600 metastatic melanoma patients treated with BRAF inhibitors. Pigment Cell Melanoma Res 29 (6): 679-687, 2016.
- Hacker E, Olsen CM, Kvaskoff M, et al.: Histologic and Phenotypic Factors and MC1R Status Associated with BRAF(V600E), BRAF(V600K), and NRAS Mutations in a Community-Based Sample of 414 Cutaneous Melanomas. J Invest Dermatol 136 (4): 829-837, 2016.
- Thomas NE, Edmiston SN, Kanetsky PA, et al.: Associations of MC1R Genotype and Patient Phenotypes with BRAF and NRAS Mutations in Melanoma. J Invest Dermatol 137 (12): 2588-2598, 2017.
- Goldstein AM, Xiao Y, Sampson J, et al.: Rare germline variants in known melanoma susceptibility genes in familial melanoma. Hum Mol Genet 26 (24): 4886-4895, 2017.
- Yokoyama S, Woods SL, Boyle GM, et al.: A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature 480 (7375): 99-103, 2011.
- Sturm RA, Fox C, McClenahan P, et al.: Phenotypic characterization of nevus and tumor patterns in MITF E318K mutation carrier melanoma patients. J Invest Dermatol 134 (1): 141-9, 2014.
- Berwick M, MacArthur J, Orlow I, et al.: MITF E318K's effect on melanoma risk independent of, but modified by, other risk factors. Pigment Cell Melanoma Res 27 (3): 485-8, 2014.
- Ciccarese G, Dalmasso B, Bruno W, et al.: Clinical, pathological and dermoscopic phenotype of MITF p.E318K carrier cutaneous melanoma patients. J Transl Med 18 (1): 78, 2020.
- Potrony M, Puig-Butille JA, Aguilera P, et al.: Prevalence of MITF p.E318K in Patients With Melanoma Independent of the Presence of CDKN2A Causative Mutations. JAMA Dermatol 152 (4): 405-12, 2016.
- Ghiorzo P, Pastorino L, Queirolo P, et al.: Prevalence of the E318K MITF germline mutation in Italian melanoma patients: associations with histological subtypes and family cancer history. Pigment Cell Melanoma Res 26 (2): 259-62, 2013.
- Gromowski T, Masojć B, Scott RJ, et al.: Prevalence of the E318K and V320I MITF germline mutations in Polish cancer patients and multiorgan cancer risk-a population-based study. Cancer Genet 207 (4): 128-32, 2014.
- Cancer risks in BRCA2 mutation carriers. The Breast Cancer Linkage Consortium. J Natl Cancer Inst 91 (15): 1310-6, 1999.
- Moran A, O'Hara C, Khan S, et al.: Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Fam Cancer 11 (2): 235-42, 2012.
- van Asperen CJ, Brohet RM, Meijers-Heijboer EJ, et al.: Cancer risks in BRCA2 families: estimates for sites other than breast and ovary. J Med Genet 42 (9): 711-9, 2005.
- Kadouri L, Temper M, Grenader T, et al.: Absence of founder BRCA1 and BRCA2 mutations in cutaneous malignant melanoma patients of Ashkenazi origin. Fam Cancer 8 (1): 29-32, 2009.
- Gumaste PV, Penn LA, Cymerman RM, et al.: Skin cancer risk in BRCA1/2 mutation carriers. Br J Dermatol 172 (6): 1498-506, 2015.
- Johansson PA, Nathan V, Bourke LM, et al.: Evaluation of the contribution of germline variants in BRCA1 and BRCA2 to uveal and cutaneous melanoma. Melanoma Res 29 (5): 483-490, 2019.
- Bui AN, LeBoeuf NR, Nambudiri VE: Skin cancer risk in CHEK2 mutation carriers. J Eur Acad Dermatol Venereol 35 (2): 353-359, 2021.
- Weischer M, Heerfordt IM, Bojesen SE, et al.: CHEK2*1100delC and risk of malignant melanoma: Danish and German studies and meta-analysis. J Invest Dermatol 132 (2): 299-303, 2012.
- Hatton JN, Sargen MR, Frone MN, et al.: Spectrum and Incidence of Skin Cancer among Individuals with Li-Fraumeni Syndrome. J Invest Dermatol 142 (9): 2534-2537.e1, 2022.
- Mester J, Eng C: When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet 163C (2): 114-21, 2013.
- Pilarski R, Eng C: Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet 41 (5): 323-6, 2004.
- Eng C: PTEN Hamartoma Tumor Syndrome (PHTS). In: Adam MP, Feldman J, Mirzaa GM, et al., eds.: GeneReviews. University of Washington, Seattle, 1993-2023, pp. Available online. Last accessed August 3, 2023.
- Pilarski R, Burt R, Kohlman W, et al.: Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst 105 (21): 1607-16, 2013.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 1.2023. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2022. Available online with free registration. Last accessed November 8, 2023.
- Bubien V, Bonnet F, Brouste V, et al.: High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet 50 (4): 255-63, 2013.
- Tan MH, Mester JL, Ngeow J, et al.: Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 18 (2): 400-7, 2012.
- Nieuwenhuis MH, Kets CM, Murphy-Ryan M, et al.: Cancer risk and genotype-phenotype correlations in PTEN hamartoma tumor syndrome. Fam Cancer 13 (1): 57-63, 2014.
- Greene MH: The genetics of hereditary melanoma and nevi. 1998 update. Cancer 86 (11 Suppl): 2464-77, 1999.
- Olsen CM, Neale RE, Green AC, et al.: Independent validation of six melanoma risk prediction models. J Invest Dermatol 135 (5): 1377-84, 2015.
- Cho E, Rosner BA, Feskanich D, et al.: Risk factors and individual probabilities of melanoma for whites. J Clin Oncol 23 (12): 2669-75, 2005.
- Fears TR, Guerry D, Pfeiffer RM, et al.: Identifying individuals at high risk of melanoma: a practical predictor of absolute risk. J Clin Oncol 24 (22): 3590-6, 2006.
- Hampel H, Bennett RL, Buchanan A, et al.: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 17 (1): 70-87, 2015.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Melanoma: Cutaneous. Version 3.2022. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2022. Available online with free registration. Last accessed August 22, 2023.
- Kefford RF, Newton Bishop JA, Bergman W, et al.: Counseling and DNA testing for individuals perceived to be genetically predisposed to melanoma: A consensus statement of the Melanoma Genetics Consortium. J Clin Oncol 17 (10): 3245-51, 1999.
- Hansson J, Bergenmar M, Hofer PA, et al.: Monitoring of kindreds with hereditary predisposition for cutaneous melanoma and dysplastic nevus syndrome: results of a Swedish preventive program. J Clin Oncol 25 (19): 2819-24, 2007.
- Salerni G, Carrera C, Lovatto L, et al.: Benefits of total body photography and digital dermatoscopy ("two-step method of digital follow-up") in the early diagnosis of melanoma in patients at high risk for melanoma. J Am Acad Dermatol 67 (1): e17-27, 2012.
- Freedberg KA, Geller AC, Miller DR, et al.: Screening for malignant melanoma: A cost-effectiveness analysis. J Am Acad Dermatol 41 (5 Pt 1): 738-45, 1999.
- Pissa M, Lapins J, Sköldmark C, et al.: Melanoma-specific survival before and after inclusion in a familial melanoma dermatologic surveillance program in CDKN2A mutation carriers and non-carriers. J Eur Acad Dermatol Venereol 37 (2): 284-292, 2023.
- Tucker MA, Fraser MC, Goldstein AM, et al.: A natural history of melanomas and dysplastic nevi: an atlas of lesions in melanoma-prone families. Cancer 94 (12): 3192-209, 2002.
- Parker JF, Florell SR, Alexander A, et al.: Pancreatic carcinoma surveillance in patients with familial melanoma. Arch Dermatol 139 (8): 1019-25, 2003.
- Rulyak SJ, Kimmey MB, Veenstra DL, et al.: Cost-effectiveness of pancreatic cancer screening in familial pancreatic cancer kindreds. Gastrointest Endosc 57 (1): 23-9, 2003.
Syndromes Associated With Rare Types of Skin Cancer
Brooke-Spiegler Syndrome, Multiple Familial Trichoepithelioma, and Familial Cylindromatosis
Brooke-Spiegler Syndrome (BSS), familial cylindromatosis, and multiple familial trichoepithelioma (MFT) are all autosomal dominant syndromes with overlapping clinical characteristics with allelic variance. Features of BSS include multiple skin appendage tumors such as cylindromas (tumors arising in the hair follicle stem cells), trichoepitheliomas (tumors arising in the hair follicle), and spiradenomas (benign tumors arising in the sweat gland). MFT is characterized by nonmalignant skin tumors, primarily trichoepitheliomas, and familial cylindromatosis manifests predominantly as cutaneous cylindromas. Onset of tumors for these syndromes is typically in late childhood or early adolescence, suggesting a hormonal influence. There is some evidence of greater severity in females than in males. UV radiation appears to be a major initiating factor for cylindromas. Typical tumor sites for cylindromas in familial cylindromatosis are the scalp (81% of carriers), the trunk (69% of carriers), and the pubic area (42% of carriers). Other tumors that can be associated with these syndromes include parotid gland tumors, basal cell adenomas, and basal cell carcinomas. For more information about BSS, see Table 3, Basal Cell Carcinoma (BCC) Syndromes.
Because pathogenic variants in CYLD on16q12-q13 have been identified in individuals with each of these disorders, these syndromes are thought to represent different phenotypic manifestations of the same disease. Penetrance of pathogenic variants in CYLD is reported to be 60% to 100%. In one study, 85% of the BSS families, 100% of familial cylindromatosis families, and only 44% of MFT families were found to have pathogenic variants in CYLD. A second locus for MFT maps to 9p21, but the gene for this locus remains unknown.
Given the potential for progressive enlargement, the preferred approach for cylindromas is ablation while the tumors are small and easily managed. Electrosurgery or Mohs micrographic surgery may be used for therapy, although excision of large lesions may require skin grafting for closure. Trichoepitheliomas and spiradenomas typically remain smaller in size; thus, after the diagnosis is confirmed by skin biopsy, unless there is impingement on critical structures, further intervention is not required. If therapy is deemed necessary and appropriate, either electrosurgery or ablative laser therapy is a valid option. Radiotherapy is not recommended for treatment of any of these tumors because a potential for increased tumor induction.
Level of evidence: 4
Sebaceous Carcinoma
Cutaneous sebaceous neoplasms may be associated with Muir-Torre syndrome (MTS). Multiple types of sebaceous tumors including sebaceous adenomas, epitheliomas, carcinomas, and keratoacanthomas or BCCs with sebaceous differentiation have been described. A variant of Lynch syndrome/hereditary non-polyposis colorectal cancer syndrome, the MTS phenotype involves the synchronous or metachronous development of at least one cutaneous sebaceous neoplasm and at least one visceral malignancy. The visceral malignancies may be of gastrointestinal (colorectal, stomach, small bowel, liver, and bile duct) and/or genitourinary (endometrial and bladder) origin and typically demonstrate a less aggressive phenotype than non-MTS equivalent tumors. MTS, inherited in an autosomal dominant fashion with high penetrance and variable expressivity, is associated with pathogenic variants in the mismatch repair genes MLH1, MSH2, and less commonly, MSH6. In a study of 36 sebaceous lesions that included sebaceous carcinomas, sebaceous adenomas, and sebaceomas, 38.9% of lesions were missing one or more mismatch repair proteins by immunohistochemistry (IHC). Of the ten individuals with absent staining of one or more proteins, five had gene testing that confirmed a diagnosis of Lynch syndrome. This result suggests that routine screening of sebaceous lesions by IHC may be useful in identification of individuals with Lynch syndrome. One study of the incidence of skin cancer in Lynch syndrome suggests there is an increase in sebaceous carcinoma and squamous cell carcinoma in these patients.
While the commonly noted sebaceous hyperplasia has not been associated with MTS, any sebaceous lesion with atypical or difficult to classify histologic features should prompt further exploration of the patient’s family and personal medical history. Consideration should be given to referring patients with sebaceous neoplasms to medical geneticists or gastroenterologists to evaluate further for Lynch syndrome. While the diagnosis of visceral malignancy precedes that of cutaneous sebaceous neoplasms in the majority of patients, 22% of patients develop cutaneous sebaceous neoplasms first, offering an opportunity for visceral malignancy screening. Current diagnosis of MTS is based upon clinical criteria but may be supported by immunohistochemical staining for MSH2, MLH1, and MSH6 as a screening mechanism before molecular genetic analysis. Genetic counseling and testing for the patient and family members, with appropriate visceral malignancy screening regimens, should be pursued.
Level of evidence: 3
Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC)
Although cutaneous smooth muscle tumors (leiomyomas) are not themselves a form of skin cancer, multiple cutaneous leiomyomas are associated with renal cell cancer (RCC) in an inherited syndrome known as hereditary leiomyomatosis and renal cell cancer (HLRCC). HLRCC is associated with germline pathogenic variants in the FH (fumarate hydratase) gene. Cutaneous leiomyomas present as firm, pink or reddish-brown papules and nodules distributed over the trunk and extremities and, occasionally, on the face. These lesions occur at a mean age of 25 years (age range, 10–47 y) and tend to increase in size and number with age. Lesions are sensitive to light touch and/or cold temperature and are, less commonly, painful. Pain is correlated with severity of cutaneous involvement. The presence of multiple cutaneous leiomyomas is associated with HLRCC until proven otherwise and should prompt a genetic workup; a solitary leiomyoma requires careful analysis of family history. HLRCC is also associated with clinically significant uterine fibroids and aggressive renal cancers. For more information, see Hereditary Leiomyomatosis and Renal Cell Cancer.
References
- Bowen S, Gill M, Lee DA, et al.: Mutations in the CYLD gene in Brooke-Spiegler syndrome, familial cylindromatosis, and multiple familial trichoepithelioma: lack of genotype-phenotype correlation. J Invest Dermatol 124 (5): 919-20, 2005.
- Burrows NP, Jones RR, Smith NP: The clinicopathological features of familial cylindromas and trichoepitheliomas (Brooke-Spiegler syndrome): a report of two families. Clin Exp Dermatol 17 (5): 332-6, 1992.
- Rajan N, Langtry JA, Ashworth A, et al.: Tumor mapping in 2 large multigenerational families with CYLD mutations: implications for disease management and tumor induction. Arch Dermatol 145 (11): 1277-84, 2009.
- Young AL, Kellermayer R, Szigeti R, et al.: CYLD mutations underlie Brooke-Spiegler, familial cylindromatosis, and multiple familial trichoepithelioma syndromes. Clin Genet 70 (3): 246-9, 2006.
- Welch JP, Wells RS, Kerr CB: Ancell-Spiegler cylindromas (turban tumours) and Brooke-Fordyce Trichoepitheliomas: evidence for a single genetic entity. J Med Genet 5 (1): 29-35, 1968.
- Saggar S, Chernoff KA, Lodha S, et al.: CYLD mutations in familial skin appendage tumours. J Med Genet 45 (5): 298-302, 2008.
- Harada H, Hashimoto K, Ko MS: The gene for multiple familial trichoepithelioma maps to chromosome 9p21. J Invest Dermatol 107 (1): 41-3, 1996.
- Rajan N, Trainer AH, Burn J, et al.: Familial cylindromatosis and brooke-spiegler syndrome: a review of current therapeutic approaches and the surgical challenges posed by two affected families. Dermatol Surg 35 (5): 845-52, 2009.
- Schwartz RA, Torre DP: The Muir-Torre syndrome: a 25-year retrospect. J Am Acad Dermatol 33 (1): 90-104, 1995.
- Cohen PR, Kohn SR, Kurzrock R: Association of sebaceous gland tumors and internal malignancy: the Muir-Torre syndrome. Am J Med 90 (5): 606-13, 1991.
- Cerosaletti KM, Lange E, Stringham HM, et al.: Fine localization of the Nijmegen breakage syndrome gene to 8q21: evidence for a common founder haplotype. Am J Hum Genet 63 (1): 125-34, 1998.
- Mangold E, Pagenstecher C, Leister M, et al.: A genotype-phenotype correlation in HNPCC: strong predominance of msh2 mutations in 41 patients with Muir-Torre syndrome. J Med Genet 41 (7): 567-72, 2004.
- Mathiak M, Rütten A, Mangold E, et al.: Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test. Am J Surg Pathol 26 (3): 338-43, 2002.
- Mangold E, Rahner N, Friedrichs N, et al.: MSH6 mutation in Muir-Torre syndrome: could this be a rare finding? Br J Dermatol 156 (1): 158-62, 2007.
- Arnold A, Payne S, Fisher S, et al.: An individual with Muir-Torre syndrome found to have a pathogenic MSH6 gene mutation. Fam Cancer 6 (3): 317-21, 2007.
- Murphy HR, Armstrong R, Cairns D, et al.: Muir-Torre Syndrome: expanding the genotype and phenotype--a further family with a MSH6 mutation. Fam Cancer 7 (3): 255-7, 2008.
- Plocharczyk EF, Frankel WL, Hampel H, et al.: Mismatch repair protein deficiency is common in sebaceous neoplasms and suggests the importance of screening for Lynch syndrome. Am J Dermatopathol 35 (2): 191-5, 2013.
- Adan F, Crijns MB, Zandstra WSE, et al.: Cumulative risk of skin tumours in patients with Lynch syndrome. Br J Dermatol 179 (2): 522-523, 2018.
- Akhtar S, Oza KK, Khan SA, et al.: Muir-Torre syndrome: case report of a patient with concurrent jejunal and ureteral cancer and a review of the literature. J Am Acad Dermatol 41 (5 Pt 1): 681-6, 1999.
- Entius MM, Keller JJ, Drillenburg P, et al.: Microsatellite instability and expression of hMLH-1 and hMSH-2 in sebaceous gland carcinomas as markers for Muir-Torre syndrome. Clin Cancer Res 6 (5): 1784-9, 2000.
- Toro JR, Nickerson ML, Wei MH, et al.: Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 73 (1): 95-106, 2003.
Psychosocial Issues in Familial Melanoma
Introduction
This section reviews the literature examining risk reduction and early-detection behaviors in individuals with heightened risk of melanoma resulting from their family history of the disease and in individuals from hereditary families who have been tested for melanoma high-risk pathogenic variant status. The review also addresses risk perception and communication in individuals at heightened risk of melanoma.
Interest in and Uptake of Genetic Testing for Risk of Melanoma
Currently, clinical testing for CDKN2A is not recommended outside the research context because most individuals from multiple-case families will not be identified as having a pathogenic variant in this gene, and because recommendations for those testing positive do not differ for multiple-case family members who test negative, or do not pursue testing. Despite these cautions, CDKN2A testing is commercially available, and thus demand for the test will likely increase. Arguments for the availability of genetic testing include that the results of testing could provide psychological security and contribute to enhanced screening and prevention efforts for those testing positive for CDKN2A. For more information about melanoma genetic testing, see the Melanoma Risk Assessment section.
Few studies have examined motivation and interest in genetic testing for melanoma risk. In summary, the findings include the following:
- High, but not universal interest in genetic testing.
- Articulated benefits of testing among those at heightened risk.
- A relative lack of examination of potential limitations of testing or reasons to forgo testing.
In Australia, a qualitative study (N = 40) found that almost all participants with a strong family history of melanoma were interested in genetic testing. Genetic testing was favored by the participants as a means to gain information about their children's susceptibility to melanoma, to increase their understanding of their own risk, to advance melanoma research, and to provide increased motivation for sun-protective behavior.
A Dutch study examined interest in CDKN2A testing (p16-Leiden pathogenic variant). Of 510 letters sent to members of 18 p16-Leiden-positive families recruited from the Pigmented Lesions Clinic at the Leiden University Medical Center in the Netherlands, 488 individuals responded by attending clinic for physical examination; an additional 15 family members also accompanied these individuals. Of these, 403 individuals were eligible for genetic counseling. A total of 184 family members followed through with counseling, and 141 of them opted for genetic testing. After the counseling session, 94 individuals returned a completed questionnaire. Older age predicted higher interest in genetic testing; reasons for having genetic testing included learning personal risk (57%) and learning the risk of one's child carrying the pathogenic variant (69%). Most participants (88%) felt that genetic testing would contribute to diagnostics within their family. However, some individuals (40%) reported that they had not expected to receive risk information concerning pancreatic cancer, and half of the participants (49%) reported increased worry about the possibility of developing pancreatic cancer. Finally, in an Arizona qualitative study of 22 individuals with a strong family history of melanoma, none elected genetic testing even though it was provided as an option for them.
In an Australian study of 121 individuals with a strong family history of melanoma, participants completed questionnaires before genetic counseling and testing. Distress (melanoma-specific distress and general distress) levels were very low in this population. The most important predictors of distress included a personal history of melanoma, having concerns about the impact of melanoma on family, having a high information-seeking disposition (monitoring style), a perceived importance of sun exposure in causing melanoma, and not having children.
Testing in children
A systematic review described three studies that examined participants' attitudes towards performing hereditary melanoma genetic testing in minors. Most participants were in favor of testing minors for familial melanoma risk. Many participants who supported testing minors believed that genetic testing could raise awareness about melanoma risk and promote sun-protective behaviors.
Risk Awareness and Risk Reduction in Individuals at Increased Familial Risk of Melanoma
A number of studies have been conducted examining risk reduction via adoption of sun protection (including the use of sunscreen and protective clothing and shade seeking behavior) in individuals with a family history of melanoma. Overall, these studies indicate inconsistent adoption and maintenance of these behaviors. Most of these studies have been conducted with clinic-based populations that might be more prone to risk reduction and screening behaviors than those with a similar risk profile in the general population.
In terms of sun protection, in a Swedish population, 87 young adults with dysplastic nevi were surveyed, and 70% estimated their melanoma risk to be equal or lower than that of the Swedish population in general, and one third reported frequent sunbathing behavior. Another study examined 229 first-degree relatives (FDRs) referred by melanoma patients attending clinic appointments; those who were older, female, and had greater confidence in their ability to practice sun-protection were most likely to do so, but the utilization of sun-protective behavior was inconsistent. Another study in the United States examined sun-protective behavior in 100 FDRs of melanoma clinic patients and found that less than one-third of patients use sunscreen routinely when in the sun and that more regular usage was related to higher education levels, higher self-efficacy for sun protection, and higher perceived melanoma risk. Perceived severity of melanoma and response-efficacy were not related to adoption of sun-protective behaviors.
A study that focused on 68 minor children (aged 17 years or younger) of melanoma survivors demonstrated that while overall rates of sun-protective behavior were high (near 80%), the rates of sunburn were also high (49%). The authors concluded that multiple methods of sun-protective behavior are warranted in these individuals. However, in the teenage years, there were significant reductions in sun protection indicating an even greater need for intervention in this group.
Another study based in the United Kingdom examined sunburn rates in 170 individuals with a family history of melanoma compared with 140 controls matched to age, sex, and geographical location. Of those with a melanoma family history, 31% reported sunburn in the previous summer (compared with 41% of controls); melanoma families reported better sun-protection behaviors than controls overall. Across controls and those with a family history of melanoma, younger males were more likely to report recent sunburns; also, across controls and those with a family history of melanoma, those relatives with atypical mole syndrome and a belief in their ability to prevent melanoma showed better sun protection.
A large (547 families, 2,407 total individuals), retrospective, international study of a familial melanoma cohort (GenoMEL participants) examined longitudinal sun exposure and sun-protective behaviors in different birth cohorts. The researchers found that sun exposure was similar across all birth cohorts. Sunscreen use increased throughout a participant's life and was used more frequently in recent birth cohorts. There were no differences in histories of sun exposure, sunscreen use, or use of sunbeds between people who had melanoma and those who did not. However, sun-protective behaviors increased after participants were diagnosed with melanoma.
One qualitative study of 20 FDRs of melanoma patients recruited from a high-risk clinic at the University of Arizona identified perceived unmet needs for physician communication of risk status, including greater consistency in communication, education for patients concerning the importance of family history to risk status, and needs and desire for more complex advice (e.g., reapplication of sunscreen and wearing clothing with ultraviolet protection factor).
A prospective study examined interest in and 3-month behavioral and psychosocial outcomes associated with disclosure of melanoma high-risk pathogenic variant research results in 19 individuals (three CDKN2A carriers). All of the variant carriers, but only four of the noncarriers, had a family history of melanoma. Carrier status did not affect risk perception, distress, or sun-protection behaviors.
Intervention studies
A few intervention studies have targeted knowledge about melanoma, sun protection, and screening in family members of melanoma patients. In one study among siblings, participants drawn from a clinic population were randomly assigned to an intervention that included telephone messages and tailored print materials about risk reduction and screening recommendations. The usual care group received a standard physician-practice recommendation that patients notify family members about their diagnosis. The intervention group showed improvements in knowledge about melanoma, confidence in seeing a dermatologist and having a screening examination, and greater improvements in skin self-examination practices compared with control participants after 12 months; both groups showed twofold increases in physician examinations after 12 months; there was no change in sunscreen behaviors in either group.
In another study, 443 family members of melanoma patients were randomly assigned to either a generic or tailored intervention that consisted of three (untailored or tailored) print mailings and one (untailored or tailored) telephone counseling session. Overall, the tailored intervention group showed an almost twofold increase in frequency of total cutaneous skin examinations by a health care provider compared with the generic intervention. However, no differences were observed for skin self-examinations between intervention arms. In contrast to the previous study, which did not show improvements in sun protection habits, participants in this study who received the tailored intervention were significantly more likely to report improvements in sun protection habits than were those who received the generic intervention.
Screening Behaviors in Individuals at Increased Familial Risk of Melanoma
A number of studies have examined early-detection behaviors in individuals at increased risk of melanoma. In a U.S. sample of 404 siblings drawn from a clinic population of melanoma patients, only 42% of individuals had ever seen a dermatologist; 62% had engaged in skin self-examination; 27% had received a physician skin examination; and only 54% routinely used sunscreen. Female gender was related to greater sunscreen use; those older than age 50 years were more likely to have received a physician skin examination. Having a dermatologist was strongly related to all three outcomes (skin self-examination, physician examination, and sunscreen use). In a U.S. study of 229 FDRs referred by patients attending clinic, about half (55%) reported ever having a total cutaneous examination, and slightly more (71%) reported ever performing skin self-examination. Common predictors of skin examination (physician and self-examinations) included physician recommendation and low perceived barriers of screening. Interestingly, 14% of the sample had not told their primary care doctor about their sibling’s melanoma diagnosis. One U.S. study showed that half (53%) of FDRs had never received a total cutaneous screening by a physician; only 27% had received a physician recommendation to have a screening. Early detection adherence was related to the following: higher education level, more melanoma risk factors, health care provider recommendation for screening, perceived risk of melanoma, and perceived severity of melanoma. Parents of melanoma patients were less likely to have pursued screening than siblings and children. A U.S. study examined intentions to receive a physician skin examination and to perform skin self-examination among FDRs of individuals diagnosed with melanoma who had not recently engaged in skin surveillance. Predictors of intentions included both benefits and barriers to screening and family support for screening, but not knowledge of recommended screening frequency.
A cross-sectional Australian study of 120 individuals from families with a known CDKN2A pathogenic variant found that in the past 12 months, 50% reported engaging in skin self-examinations at least four times, and 43% had undergone at least one clinical skin examination. In contrast, 15% had not performed a skin self-examination in the past 12 months, and 27% had never had a clinical skin examination. Correlates of skin cancer screening behaviors included having a history of melanoma, a physician’s recommendation, and stronger behavioral intentions. Additional correlates for skin self-examination included self-efficacy, perceived efficacy of melanoma treatment, and melanoma-specific distress. Perceived risk of developing melanoma was not significantly associated with skin cancer screening behaviors.
A systematic review provided a narrative summary of behavioral outcomes (sun-protective behaviors, self-skin examination, and clinical skin examination) associated with genetic testing for inherited melanoma risk. With some differences between studies, more than one-half of studies found that an inherited pathogenic variant was associated with increased adherence (or intentions) to recommended melanoma prevention and screening. There were no decreases in sun-protective behaviors among noncarriers.
Intervention studies
A few intervention studies have targeted knowledge about melanoma, sun protection, and screening in family members of melanoma patients. In one study among siblings, participants drawn from a clinic population were randomly assigned to an intervention that included telephone messages and tailored print materials about risk reduction and screening recommendations. The usual care group received a standard physician-practice recommendation that patients notify family members about their diagnosis. The intervention group showed improvements in knowledge about melanoma, confidence in seeing a dermatologist and having a screening examination, and greater improvements in skin self-examination practices compared with control participants after 12 months; both groups showed twofold increases in physician examinations after 12 months; there was no change in sunscreen behaviors in either group.
In another study, 443 family members of melanoma patients were randomly assigned to either a generic or tailored intervention that consisted of three (untailored or tailored) print mailings and one (untailored or tailored) telephone counseling session. Overall, the tailored intervention group showed an almost twofold increase in frequency of total cutaneous skin examinations by a health care provider compared with the generic intervention. However, no differences were observed for skin self-examinations between intervention arms. In contrast to the previous study, which did not show improvements in sun protection habits, participants in this study who received the tailored intervention were significantly more likely to report improvements in sun protection habits than were those who received the generic intervention.
Psychosocial Outcomes of Genetic Counseling and Genetic Testing
A systematic review examined multiple psychosocial outcomes for high-risk people undergoing melanoma genetic testing from articles published between 1995 and 2020. Psychological outcomes included generalized anxiety and depression, melanoma-specific distress, worry about melanoma and/or pancreatic cancer, and regret about having genetic testing. Generally, distress levels (both general and melanoma-specific) were low and subclinical. Anxiety decreased over time, both for carriers and noncarriers. Cancer-specific worry had mixed results in different studies when comparing carriers with non-carriers, affected individuals with unaffected individuals, and worry over time. Studies showed that noncarriers overestimated perceived cancer risks when compared with objective estimates. Studies generally showed low levels of decisional regret about genetic testing. People tended to name more benefits of genetic testing than limitations.
References
- de Snoo FA, Bergman W, Gruis NA: Familial melanoma: a complex disorder leading to controversy on DNA testing. Fam Cancer 2 (2): 109-16, 2003.
- Kefford RF, Mann GJ: Is there a role for genetic testing in patients with melanoma? Curr Opin Oncol 15 (2): 157-61, 2003.
- Hansen CB, Wadge LM, Lowstuter K, et al.: Clinical germline genetic testing for melanoma. Lancet Oncol 5 (5): 314-9, 2004.
- Bergman W, Gruis NA: Phenotypic variation in familial melanoma: consequences for predictive DNA testing. Arch Dermatol 143 (4): 525-6, 2007.
- Bränström R, Kasparian NA, Affleck P, et al.: Perceptions of genetic research and testing among members of families with an increased risk of malignant melanoma. Eur J Cancer 48 (16): 3052-62, 2012.
- Kasparian NA, Meiser B, Butow PN, et al.: Anticipated uptake of genetic testing for familial melanoma in an Australian sample: An exploratory study. Psychooncology 16 (1): 69-78, 2007.
- de Snoo FA, Riedijk SR, van Mil AM, et al.: Genetic testing in familial melanoma: uptake and implications. Psychooncology 17 (8): 790-6, 2008.
- Loescher LJ, Crist JD, Siaki LA: Perceived intrafamily melanoma risk communication. Cancer Nurs 32 (3): 203-10, 2009 May-Jun.
- Kasparian NA, Butow PN, Meiser B, et al.: High- and average-risk individuals' beliefs about, and perceptions of, malignant melanoma: an Australian perspective. Psychooncology 17 (3): 270-9, 2008.
- Primiero CA, Yanes T, Finnane A, et al.: A Systematic Review on the Impact of Genetic Testing for Familial Melanoma II: Psychosocial Outcomes and Attitudes. Dermatology 237 (5): 816-826, 2021.
- Shuk E, Burkhalter JE, Baguer CF, et al.: Factors associated with inconsistent sun protection in first-degree relatives of melanoma survivors. Qual Health Res 22 (7): 934-45, 2012.
- Bergenmar M, Brandberg Y: Sunbathing and sun-protection behaviors and attitudes of young Swedish adults with hereditary risk for malignant melanoma. Cancer Nurs 24 (5): 341-50, 2001.
- Manne S, Fasanella N, Connors J, et al.: Sun protection and skin surveillance practices among relatives of patients with malignant melanoma: prevalence and predictors. Prev Med 39 (1): 36-47, 2004.
- Azzarello LM, Dessureault S, Jacobsen PB: Sun-protective behavior among individuals with a family history of melanoma. Cancer Epidemiol Biomarkers Prev 15 (1): 142-5, 2006.
- Glenn BA, Bastani R, Chang LC, et al.: Sun protection practices among children with a family history of melanoma: a pilot study. J Cancer Educ 27 (4): 731-7, 2012.
- Newton Bishop JA, Gruis NA: Genetics: what advice for patients who present with a family history of melanoma? Semin Oncol 34 (6): 452-9, 2007.
- Lacson JCA, Zamani SA, Froes LAR, et al.: Birth cohort-specific trends of sun-related behaviors among individuals from an international consortium of melanoma-prone families. BMC Public Health 21 (1): 692, 2021.
- Loescher LJ, Crist JD, Cranmer L, et al.: Melanoma high-risk families' perceived health care provider risk communication. J Cancer Educ 24 (4): 301-7, 2009.
- Christensen KD, Roberts JS, Shalowitz DI, et al.: Disclosing individual CDKN2A research results to melanoma survivors: interest, impact, and demands on researchers. Cancer Epidemiol Biomarkers Prev 20 (3): 522-9, 2011.
- Geller AC, Emmons KM, Brooks DR, et al.: A randomized trial to improve early detection and prevention practices among siblings of melanoma patients. Cancer 107 (4): 806-14, 2006.
- Manne S, Jacobsen PB, Ming ME, et al.: Tailored versus generic interventions for skin cancer risk reduction for family members of melanoma patients. Health Psychol 29 (6): 583-93, 2010.
- Geller AC, Emmons K, Brooks DR, et al.: Skin cancer prevention and detection practices among siblings of patients with melanoma. J Am Acad Dermatol 49 (4): 631-8, 2003.
- Azzarello LM, Jacobsen PB: Factors influencing participation in cutaneous screening among individuals with a family history of melanoma. J Am Acad Dermatol 56 (3): 398-406, 2007.
- Coups EJ, Manne SL, Jacobsen PB, et al.: Skin surveillance intentions among family members of patients with melanoma. BMC Public Health 11: 866, 2011.
- Kasparian NA, McLoone JK, Meiser B, et al.: Skin cancer screening behaviours among individuals with a strong family history of malignant melanoma. Br J Cancer 103 (10): 1502-9, 2010.
- Primiero CA, Yanes T, Finnane A, et al.: A Systematic Review on the Impact of Genetic Testing for Familial Melanoma I: Primary and Secondary Preventative Behaviours. Dermatology 237 (5): 806-815, 2021.
Latest Updates to This Summary (11/06/2023)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Squamous Cell Carcinoma
Added text to state that oculocutaneous albinism type 4 (OCA4) is thought to be similar to other types of albinism. Also added text to state that skin cancer was not identified in a study of 30 individuals with OCA4 (cited Moreno-Artero et al. as reference 90).
Added text to state that a German study found an epidermolysis bullosa (EB) incidence of 45.1 per million live births (cited Has et al. as reference 130). Also added text to state that a study from England and Wales found a much higher EB incidence of 67.8 per million population (cited Petrof et al. as reference 131).
Added text to state that studies from the Netherlands, Germany, and England/Wales showed a range of dystrophic EB (DEB) incidence from 14.1 to 26.1 per million live births.
Added text to state that an epidemiological study in Germany found that the mean age of death was 28.03 years in patients with DEB.
Added text to state that incidence of junctional EB (JEB) in England and Wales was similar to those from the Netherlands and the U.S., but a study from Germany reported a higher incidence of JEB.
Melanoma
Added text to state that surveillance has been shown to detect melanomas at an earlier stage than melanomas diagnosed before the identification of a CDKN2A pathogenic variant (cited Pissa et al. as reference 230).
This summary is written and maintained by the PDQ Cancer Genetics Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the genetics of skin cancer. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Cancer Genetics Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Genetics of Skin Cancer are:
- Kathleen A. Calzone, PhD, RN, AGN-BC, FAAN (National Cancer Institute)
- Joanne Marie Jeter, MD (City of Hope)
- Kelly Carter Nelson, MD, FAAD (M.D. Anderson Cancer Center at University of Texas)
- Suzanne C. O'Neill, PhD (Georgetown University)
- Susan K. Peterson, PhD, MPH (University of Texas, M.D. Anderson Cancer Center)
- John M. Quillin, PhD, MPH, MS (Virginia Commonwealth University)
- Charite Ricker, MS, CGC (University of Southern California)
- Amanda Ewart Toland, PhD, FACMG (The Ohio State University)
- Catharine Wang, PhD, MSc (Boston University School of Public Health)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Cancer Genetics Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Cancer Genetics Editorial Board. PDQ Genetics of Skin Cancer. Bethesda, MD: National Cancer Institute. Updated
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
The information in these summaries should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.