National Cancer Institute
Post Date: Jun 24, 2024
This Childhood Cancer Genomics summary provides a brief synopsis of current knowledge about the genomic landscape of specific childhood cancers. Get detailed information about various genetic alterations and precision medicine concepts in childhood cancers in this summary for clinicians.
Childhood Cancer Genomics
General Information About Childhood Cancer Genomics
Research teams from around the world have made remarkable progress in the past decade in elucidating the genomic landscape of most types of childhood cancer. A decade ago it was possible to hope that targetable oncogenes, such as activated tyrosine kinases, might be identified in a high percentage of childhood cancers. However, it is now clear that the genomic landscape of childhood cancer is highly varied, and in many cases is quite distinctive from that of the common adult cancers.
There are examples of genomic lesions that have provided immediate therapeutic direction, including the following:
- NPM::ALK fusion genes associated with anaplastic large cell lymphoma cases.
- ALK point variants associated with a subset of neuroblastoma cases.
- BRAF and other kinase genomic alterations associated with subsets of pediatric glioma cases.
- Hedgehog pathway variants associated with a subset of medulloblastoma cases.
- ABL family genes activated by translocation in a subset of acute lymphoblastic leukemia (ALL) cases.
For some cancers, the genomic findings have been highly illuminating in the identification of genomically defined subsets of patients within histologies that have distinctive biological features and distinctive clinical characteristics (particularly in terms of prognosis). In some instances, identification of these subtypes has resulted in early clinical translation as exemplified by the WNT subgroup of medulloblastoma. Because of its excellent outcome, the WNT subgroup will be studied separately in future medulloblastoma clinical trials so that reductions in therapy can be evaluated with the goal of maintaining favorable outcome while reducing long-term morbidity. However, the prognostic significance of the recurring genomic lesions for some other cancers remains to be defined.
A key finding from genomic studies is the extent to which the molecular characteristics of childhood cancers correlate with their tissue (cell) of origin. As with most adult cancers, variants in childhood cancers do not arise at random, but rather are linked in specific constellations to disease categories. A few examples include the following:
- The presence of H3.3 and H3.1 K27M variants almost exclusively among pediatric midline high-grade gliomas.
- The loss of SMARCB1 in rhabdoid tumors.
- The presence of RELA translocations in supratentorial ependymomas.
- The presence of specific fusion proteins in different pediatric sarcomas.
Another theme across multiple childhood cancers is the contribution of variants of genes involved in normal development of the tissue of origin of the cancer and the contribution of genes involved in epigenomic regulation.
Structural variations play an important role for many childhood cancers. Translocations resulting in oncogenic fusion genes or overexpression of oncogenes play a central role, particularly for the leukemias and sarcomas. However, for other childhood cancers that are primarily characterized by structural variations, functional fusion genes are not produced. Mechanisms by which these recurring structural variations have oncogenic effects have been identified for osteosarcoma (translocations confined to the first intron of TP53) and medulloblastoma (structural variants juxtapose GFI1 or GFI1B coding sequences proximal to active enhancer elements leading to transcriptional activation [enhancer hijacking]). However, the oncogenic mechanisms of action for recurring structural variations of other childhood cancers (e.g., the segmental chromosomal alterations in neuroblastoma) need to be elucidated.
Understanding of the contribution of germline variants to childhood cancer etiology is being advanced by the application of whole-genome and exome sequencing to cohorts of children with cancer. Estimates for rates of pathogenic germline variants approaching 10% have emerged from studies applying these sequencing methods to childhood cancer cohorts. In some cases, the pathogenic germline variants are clearly contributory to the patient’s cancer (e.g., TP53 variants arising in the context of Li-Fraumeni syndrome), whereas in other cases the contribution of the germline variant to the patient’s cancer is less clear (e.g., variants in adult cancer predisposition genes such as BRCA1 and BRCA2 that have an undefined role in childhood cancer predisposition). The frequency of germline variants varies by tumor type (e.g., lower for neuroblastoma and higher for osteosarcoma), and many of the identified germline variants fit into known predisposition syndromes (e.g., DICER1 for pleuropulmonary blastoma, SMARCB1 and SMARCA4 for rhabdoid tumor and small cell ovarian cancer, TP53 for adrenocortical carcinoma and Li-Fraumeni syndrome cancers, RB1 for retinoblastoma, etc.). The germline contribution to the development of specific cancers is discussed in the disease-specific sections that follow.
Each section of this document is meant to provide readers with a brief summary of current knowledge about the genomic landscape of specific childhood cancers, an understanding that is critical in considering how to apply precision medicine concepts to childhood cancers.
References
- Northcott PA, Lee C, Zichner T, et al.: Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511 (7510): 428-34, 2014.
- Chen X, Bahrami A, Pappo A, et al.: Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep 7 (1): 104-12, 2014.
- Mody RJ, Wu YM, Lonigro RJ, et al.: Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. JAMA 314 (9): 913-25, 2015.
- Parsons DW, Roy A, Yang Y, et al.: Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid Tumors. JAMA Oncol 2 (5): 616-624, 2016.
- Zhang J, Walsh MF, Wu G, et al.: Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med 373 (24): 2336-46, 2015.
Leukemias
Acute Lymphoblastic Leukemia (ALL)
Genomics of childhood ALL
The genomics of childhood acute lymphoblastic leukemia (ALL) has been extensively investigated, and multiple distinctive subtypes have been defined on the basis of cytogenetic and molecular characterizations, each with its own pattern of clinical and prognostic characteristics. The discussion of the genomics of childhood ALL below is divided into three sections: the genomic alterations associated with B-ALL, followed by the genomic alterations associated with T-ALL and mixed phenotype acute leukemia (MPAL). Figures 1, 2, and 4 illustrate the distribution of B-ALL (stratified by National Cancer Institute [NCI] standard- and high-risk B-ALL) and T-ALL cases by cytogenetic/molecular subtypes.
Throughout this section, the percentages of genomic subtypes from among all B-ALL and T-ALL cases are derived primarily from a report describing the genomic characterization of patients treated on several Children's Oncology Group (COG) and St. Jude Children's Research Hospital (SJCRH) clinical trials. Percentages by subtype are presented for NCI standard-risk and NCI high-risk patients with B-ALL (up to age 18 years).
B-ALL cytogenetics/genomics
B-ALL is typified by genomic alterations that include: 1) gene fusions that lead to aberrant activity of transcription factors, 2) chromosomal gains and losses (e.g., hyperdiploidy or hypodiploidy), and 3) alterations leading to activation of tyrosine kinase genes. Figures 1 and 2 illustrate the distribution of NCI standard-risk and high-risk B-ALL cases by 23 cytogenetic/molecular subtypes. The two most common subtypes (hyperdiploid and ETV6::RUNX1 fusion) together account for approximately 60% of NCI standard-risk B-ALL cases, but only approximately 25% of NCI high-risk cases. Most other subtypes are much less common, with most occurring at frequencies less than 2% to 3% of B-ALL cases. The molecular and clinical characteristics of some of the subtypes are discussed below.
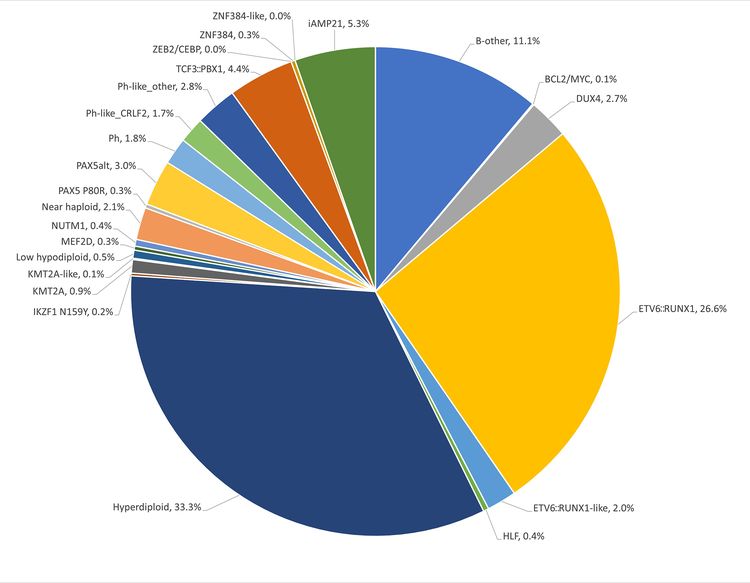 Figure 1. Genomic subtypes and frequencies of NCI standard-risk B-ALL. The figure represents data from 1,126 children diagnosed with NCI standard-risk B-ALL (aged 1–9 years and WBC <50,000/µL) and enrolled in St. Jude Children’s Research Hospital or Children’s Oncology Group clinical trials. Adapted from Supplemental Table 2 of Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 54: 1376-1389, 2022.
Figure 1. Genomic subtypes and frequencies of NCI standard-risk B-ALL. The figure represents data from 1,126 children diagnosed with NCI standard-risk B-ALL (aged 1–9 years and WBC <50,000/µL) and enrolled in St. Jude Children’s Research Hospital or Children’s Oncology Group clinical trials. Adapted from Supplemental Table 2 of Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 54: 1376-1389, 2022.
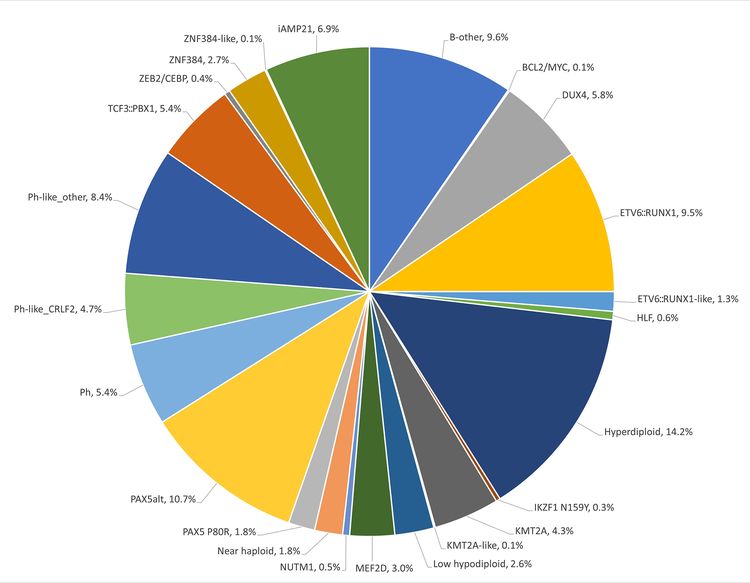 Figure 2. Genomic subtypes and frequencies of NCI high-risk B-ALL. The figure represents data from 1,084 children diagnosed with NCI high-risk B-ALL (aged 1–18 years and WBC >50,000/µL) and enrolled in St. Jude Children’s Research Hospital or Children’s Oncology Group clinical trials. Adapted from Supplemental Table 2 of Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 54: 1376-1389, 2022.
Figure 2. Genomic subtypes and frequencies of NCI high-risk B-ALL. The figure represents data from 1,084 children diagnosed with NCI high-risk B-ALL (aged 1–18 years and WBC >50,000/µL) and enrolled in St. Jude Children’s Research Hospital or Children’s Oncology Group clinical trials. Adapted from Supplemental Table 2 of Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 54: 1376-1389, 2022.
The genomic landscape of B-ALL is characterized by a range of genomic alterations that disrupt normal B-cell development and, in some cases, by variants in genes that provide a proliferation signal (e.g., activating variants in RAS family genes or variants/translocations leading to kinase pathway signaling). Genomic alterations leading to blockage of B-cell development include translocations (e.g., TCF3::PBX1 and ETV6::RUNX1 fusions), point variants (e.g., IKZF1 and PAX5), and intragenic/intergenic deletions (e.g., IKZF1, PAX5, EBF, and ERG).
The genomic alterations in B-ALL tend not to occur at random, but rather to cluster within subtypes that can be delineated by biological characteristics such as their gene expression profiles. Cases with recurring chromosomal translocations (e.g., TCF3::PBX1 and ETV6::RUNX1 fusions and KMT2A-rearranged ALL) have distinctive biological features and illustrate this point, as do the examples below of specific genomic alterations within unique biological subtypes:
- IKZF1 deletions and variants are most commonly observed within cases of BCR::ABL1 ALL and BCR::ABL1-like ALL.
- Intragenic ERG deletions occur within a distinctive subtype characterized by gene rearrangements involving DUX4.
- TP53 variants, often germline, occur at high frequency in patients with low hypodiploid ALL with 32 to 39 chromosomes. TP53 variants are uncommon in other patients with B-ALL.
Activating point variants in kinase genes are uncommon in high-risk B-ALL. JAK genes are the primary kinases that are found to be altered. These variants are generally observed in patients with BCR::ABL1-like ALL who have CRLF2 abnormalities, although JAK2 variants are also observed in approximately 25% of children with Down syndrome and ALL, occurring exclusively in cases with CRLF2 gene rearrangements. Several kinase genes and cytokine receptor genes are activated by translocations, as described below in the discussion of BCR::ABL1 ALL and BCR::ABL1-like ALL. FLT3 variants occur in a minority of cases (approximately 10%) of hyperdiploid ALL and KMT2A-rearranged ALL, and are rare in other subtypes.
Understanding of the genomics of B-ALL at relapse is less advanced than the understanding of ALL genomics at diagnosis. Childhood ALL is often polyclonal at diagnosis and under the selective influence of therapy, some clones may be extinguished and new clones with distinctive genomic profiles may arise. However, molecular subtype–defining lesions such as translocations and aneuploidy are almost always retained at relapse. Of particular importance are new variants that arise at relapse that may be selected by specific components of therapy. As an example, variants in NT5C2 are not found at diagnosis, whereas specific variants in NT5C2 were observed in 7 of 44 (16%) and 9 of 20 (45%) cases of B-ALL with early relapse that were evaluated for this variant in two studies. NT5C2 variants are uncommon in patients with late relapse, and they appear to induce resistance to mercaptopurine and thioguanine. Another gene that is found altered only at relapse is PRSP1, a gene involved in purine biosynthesis. Variants were observed in 13.0% of a Chinese cohort and 2.7% of a German cohort, and were observed in patients with on-treatment relapses. The PRSP1 variants observed in relapsed cases induce resistance to thiopurines in leukemia cell lines. CREBBP variants are also enriched at relapse and appear to be associated with increased resistance to glucocorticoids. With increased understanding of the genomics of relapse, it may be possible to tailor upfront therapy to avoid relapse or detect resistance-inducing variants early and intervene before a frank relapse.
Several recurrent chromosomal abnormalities have been shown to have prognostic significance, especially in B-ALL. Some chromosomal alterations are associated with more favorable outcomes, such as favorable trisomies (51–65 chromosomes) and the ETV6::RUNX1 fusion.[Level of evidence B4] Other alterations historically have been associated with a poorer prognosis, including the BCR::ABL1 fusion (Philadelphia chromosome–positive [Ph+]; t(9;22)(q34;q11.2)), rearrangements of the KMT2A gene, hypodiploidy, and intrachromosomal amplification of the RUNX1 gene (iAMP21).
In recognition of the clinical significance of many of these genomic alterations, the 5th edition revision of the World Health Organization Classification of Haematolymphoid Tumours lists the following entities for B-ALL:
- B-lymphoblastic leukemia/lymphoma, NOS.
- B-lymphoblastic leukemia/lymphoma with high hyperdiploidy.
- B-lymphoblastic leukemia/lymphoma with hypodiploidy.
- B-lymphoblastic leukemia/lymphoma with iAMP21.
- B-lymphoblastic leukemia/lymphoma with BCR::ABL1 fusion.
- B-lymphoblastic leukemia/lymphoma with BCR::ABL1-like features.
- B-lymphoblastic leukemia/lymphoma with KMT2A rearrangement.
- B-lymphoblastic leukemia/lymphoma with ETV6::RUNX1 fusion.
- B-lymphoblastic leukemia/lymphoma with ETV6::RUNX1-like features.
- B-lymphoblastic leukemia/lymphoma with TCF3::PBX1 fusion.
- B-lymphoblastic leukemia/lymphoma with IGH::IL3 fusion.
- B-lymphoblastic leukemia/lymphoma with TCF3::HLF fusion.
- B-lymphoblastic leukemia/lymphoma with other defined genetic abnormalities.
The category of B-ALL with other defined genetic abnormalities includes potential novel entities, including B-ALL with DUX4, MEF2D, ZNF384 or NUTM1 rearrangements; B-ALL with IG::MYC fusions; and B-ALL with PAX5alt or PAX5 p.P80R (NP_057953.1) abnormalities.
These and other chromosomal and genomic abnormalities for childhood ALL are described below.
- Chromosome number.
- High hyperdiploidy (51–65 chromosomes).
High hyperdiploidy, defined as 51 to 65 chromosomes per cell or a DNA index greater than 1.16, occurs in approximately 33% of NCI standard-risk and 14% of NCI high-risk pediatric B-ALL cases. Hyperdiploidy can be evaluated by measuring the DNA content of cells (DNA index) or by karyotyping. In cases with a normal karyotype or in which standard cytogenetic analysis was unsuccessful, interphase fluorescence in situ hybridization (FISH) may detect hidden hyperdiploidy.
High hyperdiploidy generally occurs in cases with clinically favorable prognostic factors (patients aged 1 to <10 years with a low white blood cell [WBC] count) and is an independent favorable prognostic factor. Within the hyperdiploid range of 51 to 65 chromosomes, patients with higher modal numbers (58–66) appeared to have a better prognosis in one study. Hyperdiploid leukemia cells are particularly susceptible to undergoing apoptosis and accumulate higher levels of methotrexate and its active polyglutamate metabolites, which may explain the favorable outcome commonly observed in these cases.
While the overall outcome of patients with high hyperdiploidy is considered to be favorable, factors such as age, WBC count, specific trisomies, and early response to treatment have been shown to modify its prognostic significance.
Multiple reports have described the prognostic significance of specific chromosome trisomies among children with hyperdiploid B-ALL.
- A study combining experience from the Children's Cancer Group and the Pediatric Oncology Group (POG) found that patients with trisomies of chromosomes 4, 10, and 17 (triple trisomies) have a particularly favorable outcome.; [Level of evidence B4]
- A report using POG data found that NCI standard-risk patients with trisomies of 4 and 10, without regard to chromosome 17 status, have an excellent prognosis. COG protocols currently use double trisomies of chromosomes 4 and 10 to define favorable hyperdiploidy.
- A retrospective analysis evaluated patients treated on two consecutive UKALL trials to identify and validate a profile to predict outcome in high hyperdiploid B-ALL. The investigators defined a good-risk group (approximately 80% of high hyperdiploidy patients) that was associated with a more favorable prognosis. Good-risk patients had either trisomies of both chromosomes 17 and 18 or trisomy of one of these two chromosomes along with absence of trisomies of chromosomes 5 and 20. All other patients were defined as poor risk and had a less favorable outcome. End-induction MRD and copy number alterations (such as IKZF1 deletion) were prognostically significant within each hyperdiploid risk group.
Chromosomal translocations may be seen with high hyperdiploidy, and in those cases, patients are more appropriately risk-classified on the basis of the prognostic significance of the translocation. For instance, in one study, 8% of patients with the BCR::ABL1 fusion also had high hyperdiploidy, and the outcome of these patients (treated without tyrosine kinase inhibitors) was inferior to that observed in non-BCR::ABL1 high hyperdiploid patients.
Certain patients with hyperdiploid ALL may have a hypodiploid clone that has doubled (masked hypodiploidy). Molecular technologies, such as single nucleotide polymorphism microarrays to detect widespread loss of heterozygosity, can be used to identify patients with masked hypodiploidy. These cases may be interpretable based on the pattern of gains and losses of specific chromosomes (hyperdiploidy with two and four copies of chromosomes rather than three copies). These patients have an unfavorable outcome, similar to those with hypodiploidy.
Near triploidy (68–80 chromosomes) and near tetraploidy (>80 chromosomes) are much less common and appear to be biologically distinct from high hyperdiploidy. Unlike high hyperdiploidy, a high proportion of near tetraploid cases harbor a cryptic ETV6::RUNX1 fusion. Near triploidy and tetraploidy were previously thought to be associated with an unfavorable prognosis, but later studies suggest that this may not be the case.
The genomic landscape of hyperdiploid ALL is characterized by variants in genes of the receptor tyrosine kinase (RTK)/RAS pathway in approximately one-half of cases. Genes encoding histone modifiers are also present in a recurring manner in a minority of cases. Analysis of variant profiles demonstrates that chromosomal gains are early events in the pathogenesis of hyperdiploid ALL and may occur in utero, while variants in RTK/RAS pathway genes are late events in leukemogenesis and are often subclonal.
- Hypodiploidy (<44 chromosomes).
B-ALL cases with fewer than the normal number of chromosomes have been subdivided in various ways, with one report stratifying on the basis of modal chromosome number into the following four groups:
- Near-haploid: 24 to 29 chromosomes (n = 46).
- Low-hypodiploid: 33 to 39 chromosomes (n = 26).
- High-hypodiploid: 40 to 43 chromosomes (n = 13).
- Near-diploid: 44 chromosomes (n = 54).
Near-haploid cases represent approximately 2% of NCI standard-risk and 2% of NCI high-risk pediatric B-ALL.
Low-hypodiploid cases represent approximately 0.5% of NCI standard-risk and 2.6% of NCI high-risk pediatric B-ALL cases.
Most patients with hypodiploidy are in the near-haploid and low-hypodiploid groups, and both of these groups have an elevated risk of treatment failure compared with nonhypodiploid cases. Patients with fewer than 44 chromosomes have a worse outcome than do patients with 44 or 45 chromosomes in their leukemic cells. Several studies have shown that patients with high minimal residual disease (MRD) (≥0.01%) after induction do very poorly, with 5-year event-free survival (EFS) rates ranging from 25% to 47%. Although hypodiploid patients with low MRD after induction fare better (5-year EFS rates, 64%–75%), their outcomes are still inferior to most children with other types of ALL.
The recurring genomic alterations of near-haploid and low-hypodiploid ALL appear to be distinctive from each other and from other types of ALL. In near-haploid ALL, alterations targeting RTK signaling, RAS signaling, and IKZF3 are common. In low-hypodiploid ALL, genetic alterations involving TP53, RB1, and IKZF2 are common. Importantly, the TP53 alterations observed in low-hypodiploid ALL are also present in nontumor cells in approximately 40% of cases, suggesting that these variants are germline and that low-hypodiploid ALL represents, in some cases, a manifestation of Li-Fraumeni syndrome. Approximately two-thirds of patients with ALL and germline pathogenic TP53 variants have hypodiploid ALL.
- High hyperdiploidy (51–65 chromosomes).
- Chromosomal translocations and gains/deletions of chromosomal segments.
- ETV6::RUNX1 fusion (t(12;21)(p13.2;q22.1)).
Fusion of the ETV6 gene on chromosome 12 to the RUNX1 gene on chromosome 21 is present in approximately 27% of NCI standard-risk and 10% of NCI high-risk pediatric B-ALL cases.
The ETV6::RUNX1 fusion produces a cryptic translocation that is detected by methods such as FISH, rather than conventional cytogenetics, and it occurs most commonly in children aged 2 to 9 years. Hispanic children with ALL have a lower incidence of ETV6::RUNX1 fusions than do White children.
Reports generally indicate favorable EFS and overall survival (OS) rates in children with the ETV6::RUNX1 fusion; however, the prognostic impact of this genetic feature is modified by the following factors:; [Level of evidence B4]
- Early response to treatment.
- NCI risk category (age and WBC count at diagnosis).
- Treatment regimen.
In one study of the treatment of newly diagnosed children with ALL, multivariate analysis of prognostic factors found age and leukocyte count, but not ETV6::RUNX1 fusion status, to be independent prognostic factors. However, another large trial only enrolled patients classified as having favorable-risk B-ALL, with low-risk clinical features, either trisomies of 4, 10, and 17 or ETV6::RUNX1 fusion, and end induction MRD less than 0.01%. Patients had a 5-year continuous complete remission rate of 93.7% and a 6-year OS rate of 98.2% for patients with ETV6::RUNX1. It does not appear that the presence of secondary cytogenetic abnormalities, such as deletion of ETV6 (12p) or CDKN2A/B (9p), impacts the outcome of patients with the ETV6::RUNX1 fusion.
There is a higher frequency of late relapses in patients with ETV6::RUNX1 fusions compared with other relapsed B-ALL patients. Patients with the ETV6::RUNX1 fusion who relapse seem to have a better outcome than other relapse patients, with an especially favorable prognosis for patients who relapse more than 36 months from diagnosis. Some relapses in patients with ETV6::RUNX1 fusions may represent a new independent second hit in a persistent preleukemic clone (with the first hit being the ETV6::RUNX1 translocation).
- BCR::ABL1 fusion (t(9;22)(q34.1;q11.2); Ph+).
The BCR::ABL1 fusion leads to production of a BCR::ABL1 fusion protein with tyrosine kinase activity (see Figure 3). The BCR::ABL1 fusion occurs in approximately 2% of NCI standard-risk and 5% of NCI high-risk pediatric B-ALL cases. The BCR::ABL1 fusion is also the leukemogenic driver for chronic myeloid leukemia (CML). The most common BCR breakpoint in CML is different from the most common BCR breakpoint in ALL. The breakpoint that typifies CML produces a larger fusion protein (termed p210) than the breakpoint most commonly observed for ALL (termed p190, a smaller fusion protein).
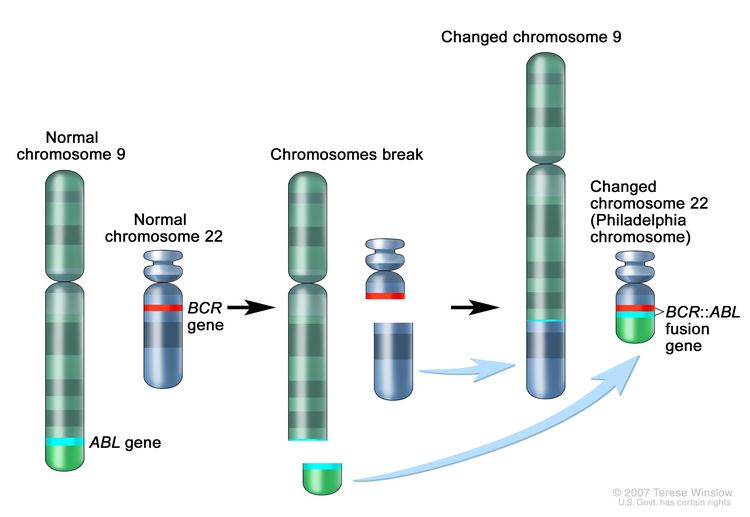 Figure 3. The Philadelphia chromosome is a translocation between the ABL1 oncogene (on the long arm of chromosome 9) and the BCR gene (on the long arm of chromosome 22), resulting in the fusion gene BCR::ABL1. BCR::ABL1 encodes an oncogenic protein with tyrosine kinase activity.
Figure 3. The Philadelphia chromosome is a translocation between the ABL1 oncogene (on the long arm of chromosome 9) and the BCR gene (on the long arm of chromosome 22), resulting in the fusion gene BCR::ABL1. BCR::ABL1 encodes an oncogenic protein with tyrosine kinase activity.Ph+ ALL is more common in older children with B-ALL and high WBC counts, with the incidence of the BCR::ABL1 fusions increasing to about 25% in young adults with ALL.
Historically, the BCR::ABL1 fusion was associated with an extremely poor prognosis (especially in those who presented with a high WBC count or had a slow early response to initial therapy), and its presence had been considered an indication for allogeneic hematopoietic stem cell transplant (HSCT) in patients in first remission. Inhibitors of the BCR::ABL1 tyrosine kinase, such as imatinib mesylate, are effective in patients with BCR::ABL1 ALL. A study by the Children's Oncology Group (COG), which used intensive chemotherapy and concurrent imatinib mesylate given daily, demonstrated a 5-year EFS rate of 70% (± 12%), which was superior to the EFS rate of historical controls in the pre-tyrosine kinase inhibitor (imatinib mesylate) era. This result eliminated the recommendation of HSCT for patients with a good early response to chemotherapy using a tyrosine kinase inhibitor.
The International Consensus Classification of acute lymphoblastic leukemia/lymphoma from 2022 divides BCR::ABL1–positive B-ALL into two subtypes: cases with lymphoid-only involvement and cases with multilineage involvement. These subtypes differ in the timing of their transformation event. A multipotent progenitor serves as the target cell of origin for BCR::ABL1–positive B-ALL with multilineage involvement, and a later progenitor is the target cell of origin for BCR::ABL1–positive B-ALL with lymphoid-only involvement.
- BCR::ABL1–positive B-ALL with lymphoid-only involvement is the predominate subtype. Only a minority of cases in children and adults have multilineage involvement (estimated at 15%–30%).
- BCR::ABL1–positive B-ALL cases with lymphoid-only involvement and cases with multilineage involvement have similar clinical presentations and immunophenotypes. In addition, both subtypes commonly have the p190 fusion protein.
- One way of distinguishing between patients with lymphoid-only and multilineage involvement is to detect the BCR::ABL1 fusion in normal non-ALL B cells, T cells, and myeloid cells.
- A second way of distinguishing between patients with lymphoid-only and multilineage involvement is to detect quantitative differences in MRD levels (typically 1 log) using measures that quantify BCR::ABL1 DNA or RNA, compared with measures based on flow cytometry, real-time quantitative polymerase chain reaction (PCR), or next-generation sequencing (NGS) quantitation of leukemia-specific immunoglobulin (IG) or T-cell receptor (TCR) rearrangements.
- For patients with lymphoid-only BCR::ABL1–positive B-ALL, MRD estimates for these methods will be correlated with each other.
- For patients with multilineage involvement BCR::ABL1–positive B-ALL, posttreatment MRD estimates based on detection of BCR::ABL1 DNA or RNA will often be higher than estimates based on flow cytometry or quantitation of leukemia-specific IG/TCR rearrangements.
- For patients with BCR::ABL1–positive B-ALL and multilineage involvement, levels of BCR::ABL1 transcripts and DNA may remain stable over time despite continued treatment with chemotherapy and tyrosine kinase inhibitors. In these situations, the persisting BCR::ABL1 DNA or RNA likely represents evidence of a residual preleukemic clone and not leukemia cells. Therefore, the term MRD is a misnomer.
- A corollary of the difference in MRD detection by methods based on BCR::ABL1 DNA or RNA detection versus MRD detection based on flow cytometry or IG/TCR rearrangements is that the latter methods provide more reliable prognostication. For example, the presence of MRD by BCR::ABL1 DNA or RNA detection in the absence of MRD detection by IG/TCR rearrangements does not confer inferior prognosis.
- Based on the limited numbers of patients studied to date, prognosis appears similar in both adults and children with lymphoid-only versus multilineage involvement BCR::ABL1–positive B-ALL.
- There are case reports of patients with multilineage involvement BCR::ABL1–positive B-ALL who relapse years from their initial diagnosis. In addition, their relapsed leukemia has the same BCR::ABL1 breakpoint as their initial leukemia, but it has a different IG/TCR rearrangement. These case reports suggest that patients with multilineage BCR::ABL1–positive B-ALL are at risk of a second leukemogenic event, leading to a second BCR::ABL1 leukemia.
- There is no evidence that a specific monitoring schedule or prolonged treatment with a tyrosine kinase inhibitor provides clinical benefit for patients with multilineage involvement BCR::ABL1–positive B-ALL who have maintained presence of BCR::ABL1 transcripts or DNA at the completion of a standard-duration course of leukemia therapy.
- KMT2A-rearranged ALL (t(v;11q23.3)).
Rearrangements involving the KMT2A gene with more than 100 translocation partner genes result in the production of fusion oncoproteins. KMT2A gene rearrangements occur in up to 80% of infants with ALL. Beyond infancy, approximately 1% of NCI standard-risk and 4% of NCI high-risk pediatric B-ALL cases have KMT2A rearrangements.
These rearrangements are generally associated with an increased risk of treatment failure, particularly in infants. The KMT2A::AFF1 fusion (t(4;11)(q21;q23)) is the most common rearrangement involving the KMT2A gene in children with ALL and occurs in approximately 1% to 2% of childhood ALL.
Patients with KMT2A::AFF1 fusions are usually infants with high WBC counts. These patients are more likely than other children with ALL to have central nervous system (CNS) disease and to have a poor response to initial therapy. While both infants and adults with the KMT2A::AFF1 fusion are at high risk of treatment failure, children with the KMT2A::AFF1 fusion appear to have a better outcome. Irrespective of the type of KMT2A gene rearrangement, infants with KMT2A-rearranged ALL have much worse event-free survival rates than non-infant pediatric patients with KMT2A-rearranged ALL.
Whole-genome sequencing has determined that cases of infant ALL with KMT2A gene rearrangements have frequent subclonal NRAS or KRAS variants and few additional genomic alterations, none of which have clear clinical significance. Deletion of the KMT2A gene has not been associated with an adverse prognosis.
Of interest, the KMT2A::MLLT1 fusion (t(11;19)(q23;p13.3)) occurs in approximately 1% of ALL cases and occurs in both early B-lineage ALL and T-ALL. Outcome for infants with the KMT2A::MLLT1 fusion is poor, but outcome appears relatively favorable in older children with T-ALL and the KMT2A::MLLT1 fusion.
- TCF3::PBX1 fusion (t(1;19)(q23;p13.3)) and TCF3::HLF fusion (t(17;19)(q22;p13)).
Fusion of the TCF3 gene on chromosome 19 to the PBX1 gene on chromosome 1 is present in approximately 4% of NCI standard-risk and 5% of NCI high-risk pediatric B-ALL cases. The TCF3::PBX1 fusion may occur as either a balanced translocation or as an unbalanced translocation and is the primary recurring genomic alteration of the pre-B–ALL immunophenotype (cytoplasmic immunoglobulin positive). Black children are relatively more likely than White children to have pre-B–ALL with the TCF3::PBX1 fusion.
The TCF3::PBX1 fusion had been associated with inferior outcome in the context of antimetabolite-based therapy, but the adverse prognostic significance was largely negated by more aggressive multiagent therapies. More specifically, in a trial conducted by St. Jude Children's Research Hospital (SJCRH) in which all patients were treated without cranial radiation, patients with the TCF3::PBX1 fusion had an overall outcome comparable to children lacking this translocation, but with a higher risk of CNS relapse and a lower rate of bone marrow relapse, suggesting that more intensive CNS therapy may be needed for these patients.
The TCF3::HLF fusion occurs in less than 1% of pediatric ALL cases. ALL with the TCF3::HLF fusion is associated with disseminated intravascular coagulation and hypercalcemia at diagnosis. Outcome is very poor for children with the TCF3::HLF fusion, with a literature review noting mortality for 20 of 21 cases reported. In addition to the TCF3::HLF fusion, the genomic landscape of this ALL subtype was characterized by deletions in genes involved in B-cell development (PAX5, BTG1, and VPREB1) and by variants in RAS pathway genes (NRAS, KRAS, and PTPN11).
- DUX4-rearranged ALL with frequent ERG deletions.
Approximately 3% of NCI standard-risk and 6% of NCI high-risk pediatric B-ALL patients have a rearrangement involving DUX4 that leads to its overexpression. East Asian ancestry was linked to an increased prevalence of DUX4-rearranged ALL (favorable). The most common rearrangement produces IGH::DUX4 fusions, with ERG::DUX4 fusions also observed. DUX4-rearranged cases show a distinctive gene expression pattern that was initially identified as being associated with focal deletions in ERG, and one-half to more than two-thirds of these cases have focal intragenic deletions involving ERG that are not observed in other ALL subtypes. ERG deletions often appear to be clonal, but using sensitive detection methodology, it appears that most cases are polyclonal. IKZF1 alterations are observed in 20% to 40% of DUX4-rearranged ALL.
ERG deletion connotes an excellent prognosis, with OS rates exceeding 90%. Even when the IZKF1 deletion is present, prognosis remains highly favorable. While patients with DUX4-rearranged ALL have an overall favorable prognosis, there is uncertainty as to whether this applies to both ERG-deleted and ERG-intact cases. In a study of 50 patients with DUX4-rearranged ALL, patients with an ERG deletion detected by genomic PCR (n = 33) had a more favorable EFS rate of approximately 90% than did patients with intact ERG (n = 17), with an EFS rate of approximately 70%.
- MEF2D-rearranged ALL.
Gene fusions involving MEF2D, a transcription factor that is expressed during B-cell development, are observed in approximately 0.3% of NCI standard-risk and 3% of NCI high-risk pediatric B-ALL cases.
Although multiple fusion partners may occur, most cases involve BCL9, which is located on chromosome 1q21, as is MEF2D. The interstitial deletion producing the MEF2D::BCL9 fusion is too small to be detected by conventional cytogenetic methods. Cases with MEF2D gene fusions show a distinctive gene expression profile, except for rare cases with MEF2D::CSFR1 that have a BCR::ABL1-like gene expression profile.
The median age at diagnosis for cases of MEF2D-rearranged ALL in studies that included both adult and pediatric patients was 12 to 14 years. For 22 children with MEF2D-rearranged ALL enrolled in a high-risk ALL clinical trial, the 5-year EFS rate was 72% (standard error, ± 10%), which was inferior to that for other patients.
- ZNF384-rearranged ALL.
ZNF384 is a transcription factor that is rearranged in approximately 0.3% of NCI standard-risk and 2.7% of NCI high-risk pediatric B-ALL cases.
East Asian ancestry was associated with an increased prevalence of ZNF384. Multiple fusion partners for ZNF384 have been reported, including ARID1B, CREBBP, EP300, SMARCA2, TAF15, and TCF3. Regardless of the fusion partner, ZNF384-rearranged ALL cases show a distinctive gene expression profile. ZNF384 rearrangement does not appear to confer independent prognostic significance. However, within the subset of patients with ZNF384 rearrangements, patients with EP300::ZNF384 fusions have lower relapse rates than patients with other ZNF384 fusion partners. The immunophenotype of B-ALL with ZNF384 rearrangement is characterized by weak or negative CD10 expression, with expression of CD13 and/or CD33 commonly observed. Cases of mixed phenotype acute leukemia (MPAL) (B/myeloid) that have ZNF384 gene fusions have been reported, and a genomic evaluation of MPAL found that ZNF384 gene fusions were present in approximately one-half of B/myeloid cases.
- NUTM1-rearranged B-ALL.
NUTM1-rearranged B-ALL is most commonly observed in infants, representing 3% to 5% of overall cases of B-ALL in this age group and approximately 20% of infant B-ALL cases lacking the KMT2A rearrangement. The frequency of NUTM1 rearrangement is lower in children after infancy (<1% of cases).
The NUTM1 gene is located on chromosome 15q14, and some cases of B-ALL with NUTM1 rearrangements show chromosome 15q aberrations, but other cases are cryptic and have no cytogenetic abnormalities. RNA sequencing, as well as break-apart FISH, can be used to detect the presence of the NUTM1 rearrangement.
The NUTM1 rearrangement appears to be associated with a favorable outcome. Among 35 infants with NUTM1-rearranged B-ALL who were treated on Interfant protocols, all patients achieved remission and no relapses were observed. For the 32 children older than 12 months with NUTM1-rearranged B-ALL, the 4-year EFS and OS rates were 92% and 100%, respectively.
- IGH::IL3 fusion (t(5;14)(q31.1;q32.3)).
This entity is included in the 2016 revision of the World Health Organization (WHO) classification of tumors of the hematopoietic and lymphoid tissues. The finding of t(5;14)(q31.1;q32.3) in patients with ALL and hypereosinophilia in the 1980s was followed by the identification of the IGH::IL3 fusion as the underlying genetic basis for the condition. The joining of the IGH locus to the promoter region of the IL3 gene leads to dysregulation of IL3 expression. Cytogenetic abnormalities in children with ALL and eosinophilia are variable, with only a subset resulting from the IGH::IL3 fusion.
The number of cases of IGH::IL3 ALL described in the published literature is too small to assess the prognostic significance of the IGH::IL3 fusion. Diagnosis of cases of IGH::IL3 ALL may be delayed because the ALL clone in the bone marrow may be small, and because it can present with hypereosinophilia in the absence of cytopenias and circulating blasts.
- Intrachromosomal amplification of chromosome 21 (iAMP21).
iAMP21 occurs in approximately 5% of NCI standard-risk and 7% of NCI high-risk pediatric B-ALL cases. iAMP21 is generally diagnosed using FISH and is defined by the presence of greater than or equal to five RUNX1 signals per cell (or ≥3 extra copies of RUNX1 on a single abnormal chromosome). iAMP21 can also be identified by chromosomal microarray analysis. Uncommonly, iAMP21 with an atypical genomic pattern (e.g., amplification of the genomic region but with less than 5 RUNX1 signals or having at least 5 RUNX1 signals with some located apart from the abnormal iAMP21-chromosome) is identified by microarray but not RUNX1 FISH. The prognostic significance of iAMP21 defined only by microarray has not been characterized.
iAMP21 is associated with older age (median, approximately 10 years), presenting WBC count of less than 50 × 109/L, a slight female preponderance, and high end-induction MRD. Analysis of variant signatures indicates that gene amplifications in iAMP21 occur later in leukemogenesis, which is in contrast to those of hyperdiploid ALL that can arise early in life and even in utero.
The United Kingdom Acute Lymphoblastic Leukaemia (UKALL) clinical trials group initially reported that the presence of iAMP21 conferred a poor prognosis in patients treated in the MRC ALL 97/99 trial (5-year EFS rate, 29%). In their subsequent trial (UKALL2003 [NCT00222612]), patients with iAMP21 were assigned to a more intensive chemotherapy regimen and had a markedly better outcome (5-year EFS rate, 78%). Similarly, the COG has reported that iAMP21 was associated with a significantly inferior outcome in NCI standard-risk patients (4-year EFS rate, 73% for iAMP21 vs. 92% in others), but not in NCI high-risk patients (4-year EFS rate, 73% vs. 80%). On multivariate analysis, iAMP21 was an independent predictor of inferior outcome only in NCI standard-risk patients. The results of the UKALL2003 and COG studies suggest that treatment of iAMP21 patients with high-risk chemotherapy regimens abrogates its adverse prognostic significance and obviates the need for HSCT in first remission.
- PAX5 alterations.
Gene expression analysis identified two distinctive ALL subsets with PAX5 genomic alterations, called PAX5alt and PAX5 p.P80R (NP_057953.1). The alterations in the PAX5alt subtype included rearrangements, sequence variants, and focal intragenic amplifications.
PAX5alt. PAX5 rearrangements have been reported to represent approximately 3% of NCI standard-risk and 11% of NCI high-risk pediatric B-ALL cases. More than 20 partner genes for PAX5 have been described, with PAX5::ETV6, the primary genomic alteration in dic(9;12)(p13;p13), being the most common gene fusion.
Intragenic amplification of PAX5 was identified in approximately 1% of B-ALL cases, and it was usually detected in cases lacking known leukemia-driver genomic alterations. Cases with PAX5 amplification show male predominance (66%), with most (55%) having NCI high-risk status. For a cohort of patients with PAX5 amplification diagnosed between 1993 and 2015, the 5-year EFS rate was 49% (95% confidence interval [CI], 36%–61%), and the OS rate was 67% (95% CI, 54%–77%), suggesting a relatively poor prognosis for patients with this B-ALL subtype.
PAX5 p.P80R (NP_057953.1). PAX5 with a p.P80R variant shows a gene expression profile distinctive from that of other cases with PAX5 alterations. Cases with PAX5 p.P80R represent approximately 0.3% of NCI standard-risk and 1.8% of NCI high-risk pediatric B-ALL. PAX5 p.P80R B-ALL appears to occur more frequently in the adolescent and young adult (AYA) and adult populations (3.1% and 4.2%, respectively).
Outcome for the pediatric patients with PAX5 p.P80R and PAX5alt treated in a COG clinical trial appears to be intermediate (5-year EFS rate, approximately 75%). PAX5alt rearrangements have also been detected in infant patients with ALL, with a reported outcome similar to KMT2A-rearranged infant ALL.
- BCR::ABL1-like (Ph-like).
BCR::ABL1-negative patients with a gene expression profile similar to BCR::ABL1-positive patients have been referred to as Ph-like, and are now referred to as BCR::ABL1-like. This occurs in 10% to 20% of pediatric B-ALL patients, increasing in frequency with age, and has been associated with an IKZF1 deletion or variant.
Retrospective analyses have indicated that patients with BCR::ABL1-like ALL have a poor prognosis. In one series, the 5-year EFS rate for NCI high-risk children and adolescents with BCR::ABL1-like ALL was 58% and 41%, respectively. While it is more frequent in older and higher-risk patients, the BCR::ABL1-like subtype has also been identified in NCI standard-risk patients. In a COG study, 13.6% of 1,023 NCI standard-risk B-ALL patients were found to have BCR::ABL1-like ALL; these patients had an inferior EFS rate compared with non–BCR::ABL1-like standard-risk patients (82% vs. 91%), although no difference in OS rate (93% vs. 96%) was noted. In one study of 40 BCR::ABL1-like patients, the adverse prognostic significance of this subtype appeared to be abrogated when patients were treated with risk-directed therapy on the basis of MRD levels.
The hallmark of BCR::ABL1-like ALL is activated kinase signaling, with approximately 35% to 50% containing CRLF2 genomic alterations and half of those cases containing concomitant JAK variants.
Many of the remaining cases of BCR::ABL1-like ALL have been noted to have a series of translocations involving tyrosine-kinase encoding ABL-class fusion genes, including ABL1, ABL2, CSF1R, and PDGFRB. Fusion proteins from these gene combinations have been noted in some cases to be transformative and have responded to tyrosine kinase inhibitors both in vitro and in vivo, suggesting potential therapeutic strategies for these patients.
BCR::ABL1-like ALL cases with non-CRLF2 genomic alterations represent approximately 3% of NCI standard-risk and 8% of NCI high-risk pediatric B-ALL cases. In a retrospective study of 122 pediatric patients (aged 1–18 years) with ABL-class fusions (all treated without tyrosine kinase inhibitors), the 5-year EFS rate was 59%, and the OS rate was 76%.
Approximately 9% of BCR::ABL1-like ALL cases result from rearrangements that lead to overexpression of a truncated erythropoietin receptor (EPOR). The C-terminal region of the receptor that is lost is the region that is altered in primary familial congenital polycythemia and that controls stability of the EPOR. The portion of the EPOR remaining is sufficient for JAK-STAT activation and for driving leukemia development. Point variants in kinase genes, aside from those in JAK1 and JAK2, are uncommon in patients with BCR::ABL1-like ALL.
CRLF2. Genomic alterations in CRLF2, a cytokine receptor gene located on the pseudoautosomal regions of the sex chromosomes, have been identified in 5% to 10% of cases of B-ALL. These alterations represent approximately 50% of cases of BCR::ABL1-like ALL. The chromosomal abnormalities that commonly lead to CRLF2 overexpression include translocations of the IGH locus (chromosome 14) to CRLF2 and interstitial deletions in pseudoautosomal regions of the sex chromosomes, resulting in a P2RY8::CRLF2 fusion. These two genomic alterations are associated with distinctive clinical and biological characteristics.
BCR::ABL1-like B-ALL with CRLF2 genomic alterations is observed in approximately 2% of NCI standard-risk and 5% of NCI high-risk pediatric B-ALL cases.
ALL with genomic alterations in CRLF2 occurs at a higher incidence in children with Hispanic or Latino genetic ancestry and American Indian genetic ancestry. In a study of 205 children with high-risk B-ALL, 18 of 51 (35.3%) Hispanic or Latino patients had CRLF2 rearrangements, compared with 11 of 154 (7.1%) cases of other declared ethnicity. In a second study, only the frequency of IGH::CRLF2 fusions was increased in Hispanic or Latino children compared with non-Hispanic or non-Latino children with B-ALL (12% vs. 2.7%). In this study, the percentage of B-ALL with P2RY8::CRLF2 fusions was approximately 6% and was not affected by ethnicity.
The P2RY8::CRLF2 fusion is observed in 70% to 75% of pediatric patients with CRLF2 genomic alterations, and it occurs in younger patients (median age, approximately 4 years vs. 14 years for patients with IGH::CRLF2). P2RY8::CRLF2 occurs not infrequently with established chromosomal abnormalities (e.g., hyperdiploidy, iAMP21, dic(9;20)), while IGH::CRLF2 is generally mutually exclusive with known cytogenetic subgroups. CRLF2 genomic alterations are observed in approximately 60% of patients with Down syndrome and ALL, with P2RY8::CRLF2 fusions being more common than IGH::CRLF2 (approximately 80%–85% vs. 15%–20%).
IGH::CRLF2 and P2RY8::CRLF2 commonly occur as an early event in B-ALL development and show clonal prevalence. However, in some cases they appear to be a late event and show subclonal prevalence. Loss of the CRLF2 genomic abnormality in some cases at relapse confirms the subclonal nature of the alteration in these cases.
CRLF2 abnormalities are strongly associated with the presence of IKZF1 deletions. Deletions of IKZF1 are more common in cases with IGH::CRLF2 fusions than in cases with P2RY8::CRLF2 fusions. Other recurring genomic alterations found in association with CRLF2 alterations include deletions in genes associated with B-cell differentiation (e.g., PAX5, BTG1, EBF1, etc.) and cell cycle control (CDKN2A), as well as genomic alterations activating JAK-STAT pathway signaling (e.g., IL7R and JAK variants).
Although the results of several retrospective studies suggest that CRLF2 abnormalities may have adverse prognostic significance in univariate analyses, most do not find this abnormality to be an independent predictor of outcome. For example, in a large European study, increased expression of CRLF2 was not associated with unfavorable outcome in multivariate analysis, while IKZF1 deletion and BCR::ABL1-like expression signatures were associated with unfavorable outcome. Controversy exists about whether the prognostic significance of CRLF2 abnormalities should be analyzed on the basis of CRLF2 overexpression or on the presence of CRLF2 genomic alterations.
- IKZF1 deletions.
IKZF1 deletions, including deletions of the entire gene and deletions of specific exons, are present in approximately 15% of B-ALL cases. Less commonly, IKZF1 can be inactivated by deleterious point variants.
Cases with IKZF1 deletions tend to occur in older children, have a higher WBC count at diagnosis, and are therefore more common in NCI high-risk patients than in NCI standard-risk patients. A high proportion of BCR::ABL1-positive cases have a deletion of IKZF1, and ALL arising in children with Down syndrome appears to have elevated rates of IKZF1 deletions. IKZF1 deletions are also common in cases with CRLF2 genomic alterations and in BCR::ABL1-like ALL cases.
Multiple reports have documented the adverse prognostic significance of an IKZF1 deletion, and most studies have reported that this deletion is an independent predictor of poor outcome in multivariate analyses.; [Level of evidence B4] However, the prognostic significance of IKZF1 may not apply equally across ALL biological subtypes, as illustrated by the apparent lack of prognostic significance in patients with ERG deletions. Similarly, the prognostic significance of the IKZF1 deletion also appeared to be minimized in a cohort of COG patients with DUX4-rearranged ALL and with ERG transcriptional dysregulation that frequently occurred by ERG deletion. The Associazione Italiana di Ematologia e Oncologia Pediatrica–Berlin-Frankfurt-Münster group reported that IKZF1 deletions were significant adverse prognostic factors only in B-ALL patients with high end-induction MRD and in whom co-occurrence of deletions of CDKN2A, CDKN2B, PAX5, or PAR1 (in the absence of ERG deletion) were identified. The poor prognosis associated with IKZF1 alterations appears to be enhanced by the concomitant finding of deletion of 22q11.22. In a study of 1,310 patients with B-ALL, approximately one-half of the patients with IKZF1 alterations also had deletion of 22q11.22. The 5-year EFS rate was 43.3% for those with both abnormalities, compared with 68.5% for patients with IKZF1 alterations and wild-type 22q11.22 (P< .001).
There are few published results of changing therapy on the basis of IKZF1 gene status. The Malaysia-Singapore group published results of two consecutive trials. In the first trial (MS2003), IKZF1 status was not considered in risk stratification, while in the subsequent trial (MS2010), IKZF1-deleted patients were excluded from the standard-risk group. Thus, more IKZF1-deleted patients in the MS2010 trial received intensified therapy. Patients with IKZF1-deleted ALL had improved outcomes in MS2010 compared with patients in MS2003, but interpretation of this observation is limited by other changes in risk stratification and therapeutic differences between the two trials.[Level of evidence B4]
In the Dutch ALL11 study, patients with IKZF1 deletions had maintenance therapy extended by 1 year, with the goal of improving outcomes. The landmark analysis demonstrated an almost threefold reduction in relapse rate and an improvement in the 2-year EFS rate (from 74.4% to 91.2%), compared with historical controls.
- MYC-rearranged ALL (8q24).
MYC gene rearrangements are a rare but recurrent finding in pediatric patients with B-ALL. Patients with rearrangements of the MYC gene and the IGH2, IGK, and IGL genes at 14q32, 2p12, and 22q11.2, respectively, have been reported. The lymphoblasts typically exhibit a precursor B-cell immunophenotype, with a French-American-British (FAB) L2 or L3 morphology, with no expression of surface immunoglobulin and kappa or lambda light chains. Concurrent MYC gene rearrangements have been observed along with additional cytogenetic rearrangements such as IGH::BCL2 or KMT2A. Patients reported in the literature have been variably treated with ALL therapy or with mature B leukemia/lymphoma treatment protocols, and the optimal treatment for this patient group remains uncertain.
- ETV6::RUNX1 fusion (t(12;21)(p13.2;q22.1)).
T-ALL cytogenetics/genomics
T-ALL is characterized by genomic alterations leading to activation of transcriptional programs related to T-cell development and by a high frequency of cases (approximately 60%) with variants in NOTCH1 and/or FBXW7 that result in activation of the NOTCH1 pathway. Cytogenetic abnormalities common in B-ALL (e.g., hyperdiploidy, 51–65 chromosomes) are rare in T-ALL.
In Figure 4 below, pediatric T-ALL cases are divided into 10 molecular subtypes based on their RNA expression and gene variant status. These cases were derived from patients enrolled in SJCRH and COG clinical trials. Each subtype is associated with dysregulation of specific genes involved in T-cell development. Within a subtype, multiple mechanisms may drive expression of the dysregulated gene. For example, for the largest subtype, TAL1, overexpression of TAL1 can result from the STIL::TAL1 fusion and a noncoding insertion variant upstream of the TAL1 locus that creates a MYB-binding site. As another example, within the HOXA group, overexpression of HOXA9 can result from multiple gene fusions, including KMT2A rearrangements, MLLT10 rearrangements, and SET::NUP214 fusions. In contrast to the molecular subtypes of B-ALL, the molecular subtypes of T-ALL are not used to define treatment interventions based on their prognostic significance or therapeutic implications.
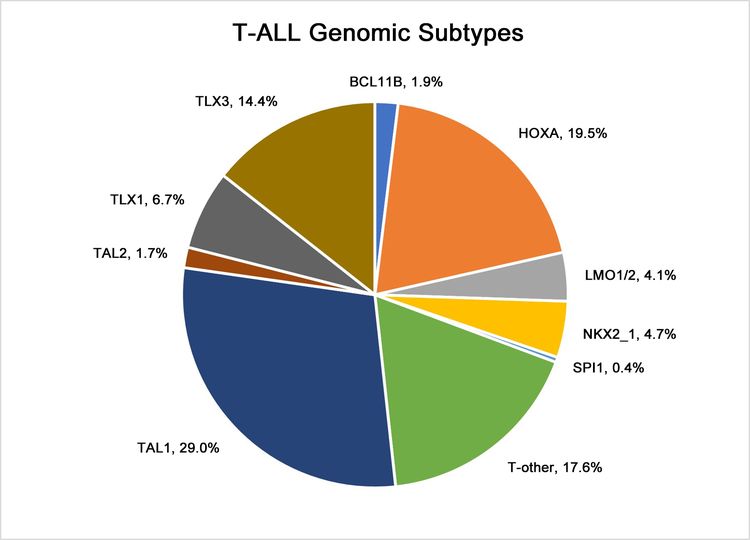 Figure 4. Genomic subtypes of T-ALL. The figure represents data from 466 children, adolescents, and young adults diagnosed with T-ALL and enrolled in St. Jude Children’s Research Hospital or Children’s Oncology Group clinical trials.
Adapted from Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 54: 1376-1389, 2022.
Figure 4. Genomic subtypes of T-ALL. The figure represents data from 466 children, adolescents, and young adults diagnosed with T-ALL and enrolled in St. Jude Children’s Research Hospital or Children’s Oncology Group clinical trials.
Adapted from Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 54: 1376-1389, 2022.
- Notch pathway signaling.
Notch pathway signaling is commonly activated by NOTCH1 and FBXW7 gene variants in T-ALL, and these are the most commonly altered genes in pediatric T-ALL. NOTCH1-activating gene variants occur in approximately 50% to 60% of T-ALL cases, and FBXW7-inactivating gene variants occur in approximately 15% of cases. Approximately 60% of T-ALL cases have Notch pathway activation by variants in at least one of these genes.
The prognostic significance of NOTCH1 and FBXW7 variants may be modulated by genomic alterations in RAS and PTEN. The French Acute Lymphoblastic Leukaemia Study Group (FRALLE) and the Group for Research on Adult Acute Lymphoblastic Leukemia reported that patients having altered NOTCH1 or FBXW7 and wild-type PTEN and RAS constituted a favorable-risk group (i.e., low-risk group), while patients with PTEN or RAS variants, regardless of NOTCH1 and FBXW7 status, have a significantly higher risk of treatment failure (i.e., high-risk group). In the FRALLE study, the 5-year disease-free survival rate was 88% for the genetic low-risk group of patients and 60% for the genetic high-risk group of patients. However, using the same criteria to define the genetic risk group, the Dana-Farber Cancer Institute consortium was unable to replicate these results. They reported a 5-year EFS rate of 86% for genetic low-risk patients and 79% for the genetic high-risk patients, a difference that was not statistically significant (P = .26).
- Chromosomal translocations.
Multiple chromosomal translocations have been identified in T-ALL that lead to deregulated expression of the target genes. These chromosome rearrangements fuse genes encoding transcription factors (e.g., TAL1, TAL2, LMO1, LMO2, LYL1, TLX1, TLX3, NKX2-I, HOXA, and MYB) to one of the T-cell receptor loci (or to other genes) and result in deregulated expression of these transcription factors in leukemia cells. These translocations are often not apparent by examining a standard karyotype, but can be identified using more sensitive screening techniques, including FISH or PCR. Variants in a noncoding region near the TAL1 gene that produce a super-enhancer upstream of TAL1 represent nontranslocation genomic alterations that can also activate TAL1 transcription to induce T-ALL.
Translocations resulting in chimeric fusion proteins are also observed in T-ALL.
- A NUP214::ABL1 fusion has been noted in 4% to 6% of T-ALL cases and is observed in both adults and children, with a male predominance. The fusion is cytogenetically cryptic and is seen in FISH on amplified episomes or, more rarely, as a small homogeneous staining region. T-ALL may also uncommonly show ABL1 fusion proteins with other gene partners (e.g., ETV6, BCR, and EML1). ABL tyrosine kinase inhibitors, such as imatinib or dasatinib, may demonstrate therapeutic benefits in this T-ALL subtype, although clinical experience with this strategy is very limited.
- Gene fusions involving SPI1 (encoding the transcription factor PU.1) were reported in 4% of Japanese children with T-ALL. Fusion partners included STMN1 and TCF7. T-ALL cases with SPI1 fusions had a particularly poor prognosis; six of seven affected individuals died within 3 years of diagnosis of early relapse.
- BCL11B is a zinc finger transcription factor that plays a dual role as a transcription activator and repressor. It is known to play a critical role in T-cell differentiation. In T-ALL, the BCL11B gene is involved in a t(5;14)(q35;q32) translocation where a distal BCL11B enhancer drives aberrant expression of TLX3 (or NKX2-5). In the process of donating its enhancer, one allele of BCL11B is inactivated. However, the resulting haploinsufficient state itself may also play a role in tumor pathogenesis. The role of BCL11B as a tumor suppressor gene is supported by the finding that about 16% of patients have T-ALL that harbors deletions or missense variants. As described in the sections for early T-cell precursor (ETP) and T/myeloid mixed phenotype acute leukemia (T/M MPAL), BCL11B may also be leukemogenic through overexpression.
- Other recurring gene fusions in T-ALL patients include those involving MLLT10, KMT2A, NUP214, and NUP98.
- Ploidy.
- Recurrent abnormalities in chromosome number are much less common in T-ALL than in B-ALL. One study included 2,250 pediatric patients with T-ALL who were treated in Associazione Italiana di Ematologia e Oncologia Pediatrica/Berlin-Frankfurt-Münster protocols. The study found that near tetraploidy (DNA index, 1.79–2.28 or 81–103 chromosomes), observed in 1.4% of patients, was associated with favorable disease features and outcomes.
Early T-cell precursor (ETP) ALL cytogenetics/genomics
Detailed molecular characterization of ETP ALL showed this entity to be highly heterogeneous at the molecular level, with no single gene affected by variant or copy number alteration in more than one-third of cases. Compared with other T-ALL cases, the ETP group had a lower rate of NOTCH1 variants and significantly higher frequencies of alterations in genes regulating cytokine receptors and RAS signaling, hematopoietic development, and histone modification. The transcriptional profile of ETP ALL shows similarities to that of normal hematopoietic stem cells and myeloid leukemia stem cells.
Studies have found that the absence of biallelic deletion of the TCR-gamma locus (ABD), as detected by comparative genomic hybridization and/or quantitative DNA-PCR, was associated with early treatment failure in patients with T-ALL. ABD is characteristic of early thymic precursor cells, and many of the T-ALL patients with ABD have an immunophenotype consistent with the diagnosis of ETP phenotype.
Allele-specific, generally high expression of BCL11B plays an oncogenic role in a subset of cases identified as ETP ALL (7 of 58 in one study) as well as in up to 30% to 40% of lineage ambiguous leukemia T/M mixed phenotype acute leukemia (T/M MPAL). The dysregulated expression of BCL11B can occur by multiple mechanisms.
- One such alteration is t(2;14)(q22;q32), which produces an in-frame ZEB2::BCL11B fusion gene.
- Other structural variants leading to allele-specific deregulated BCL11B expression include structural variants that juxtapose regulatory sequences of active genes (e.g., ARID1B [chromosome 6], BENC [chromosome 7], and CDK6 [chromosome 7]) upstream or downstream of the BCL11B locus leading to aberrant expression in a process called enhancer hijacking.
- Finally, in about 20% of cases with deregulated BCL11B expression, a translocation cannot be identified. In many such cases, amplification of a downstream enhancer, BCL11B enhancer tandem amplification (BETA), leads to BCL11B promoter driven transcription.
- There is a high prevalence of FLT3 alterations and JAK/STAT activation in acute leukemias driven by genomic alterations leading to BCL11B expression.
Mixed phenotype acute leukemia (MPAL) cytogenetics/genomics
For acute leukemias of ambiguous lineage, the WHO classification system is summarized in Table 1. The criteria for lineage assignment for a diagnosis of MPAL are provided in Table 2.
| Condition | Definition |
|---|---|
| MPAL = mixed phenotype acute leukemia; NOS = not otherwise specified. | |
| aAdapted from Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009. Obtained from Haematologica/the Hematology Journal website http://www.haematologica.org. | |
| Acute undifferentiated leukemia | Acute leukemia that does not express any marker considered specific for either lymphoid or myeloid lineage |
| MPAL with BCR::ABL1 (t(9;22)(q34;q11.2)) | Acute leukemia meeting the diagnostic criteria for MPAL in which the blasts also have the (9;22) translocation or the BCR::ABL1 rearrangement |
| MPAL with KMT2A (t(v;11q23)) | Acute leukemia meeting the diagnostic criteria for MPAL in which the blasts also have a translocation involving the KMT2A gene |
| MPAL, B/myeloid, NOS (B/M MPAL) | Acute leukemia meeting the diagnostic criteria for assignment to both B and myeloid lineage, in which the blasts lack genetic abnormalities involving BCR::ABL1 or KMT2A |
| MPAL, T/myeloid, NOS (T/M MPAL) | Acute leukemia meeting the diagnostic criteria for assignment to both T and myeloid lineage, in which the blasts lack genetic abnormalities involving BCR::ABL1 or KMT2A |
| MPAL, B/myeloid, NOS—rare types | Acute leukemia meeting the diagnostic criteria for assignment to both B and T lineage |
| Other ambiguous lineage leukemias | Natural killer–cell lymphoblastic leukemia/lymphoma |
| Lineage | Criteria |
|---|---|
| aAdapted from Arber et al. | |
| bStrong defined as equal to or brighter than the normal B or T cells in the sample. | |
| Myeloid lineage | Myeloperoxidase (flow cytometry, immunohistochemistry, or cytochemistry); or monocytic differentiation (at least two of the following: nonspecific esterase cytochemistry, CD11c, CD14, CD64, lysozyme) |
| T lineage | Strongb cytoplasmic CD3 (with antibodies to CD3 epsilon chain); or surface CD3 |
| B lineage | Strongb CD19 with at least one of the following strongly expressed: CD79a, cytoplasmic CD22, or CD10; or weak CD19 with at least two of the following strongly expressed: CD79a, cytoplasmic CD22, or CD10 |
The classification system for MPAL includes two entities that are defined by their primary molecular alteration: MPAL with BCR::ABL1 translocation and MPAL with KMT2A rearrangement. The genomic alterations associated with the MPAL, B/myeloid, NOS (B/M MPAL) and MPAL, T/myeloid, NOS (T/M MPAL) entities are distinctive, as described below:
- B/M MPAL.
- Among 115 MPAL cases for which genomic characterization was performed, 35 (30%) were B/M MPAL. There were an additional 16 MPAL cases (14%) with KMT2A rearrangements, 15 of whom showed a B/myeloid immunophenotype.
- Approximately one-half of B/M MPAL cases had rearrangements of ZNF384 with recurrent fusion partners, including TCF3 and EP300. These cases had gene expression profiles indistinguishable from B-ALL cases with ZNF384 rearrangements.
- Approximately two-thirds of B/M MPAL cases had RAS pathway alterations, with NRAS and PTPN11 being the most commonly altered genes.
- Genes encoding epigenetic regulators (e.g., MLLT3, KDM6A, EP300, and CREBBP) are altered in approximately two-thirds of B/M MPAL cases.
- T/M MPAL.
- Among 115 MPAL cases for which genomic characterization was performed, 49 (43%) were T/M MPAL. The genomic features of the T/M MPAL cases shared commonalities with those of ETP ALL, suggesting that T/M MPAL and ETP ALL are similar entities along the spectrum of immature leukemias.
- Compared with T-ALL, T/M MPAL showed a lower rate of alterations in the core T-ALL transcription factors (TAL1, TAL2, TLX1, TLX3, LMO1, LMO2, NKX2-1, HOXA10, and LYL1) (63% vs. 16%, respectively). A similar lower rate was also observed for ETP ALL.
- CDKN2A, CDKN2B, and NOTCH1 variants, which are present in approximately two-thirds of T-ALL cases, were much less common in T/M MPAL cases. By contrast, WT1 variants occurred in approximately 40% of T/M MPAL, but in less than 10% of T-ALL cases.
- One-third of T/M MPAL cases have genomic alterations associated with BCL11B that lead to allele-specific, generally high expression of BCL11B.
- One such alteration is t(2;14)(q22;q32), which produces an in-frame ZEB2::BCL11B fusion gene that leads to deregulated expression of BCL11B.
- Other alterations leading to allele-specific deregulated BCL11B expression include structural variants that juxtapose regulatory sequences of active genes (e.g., ARID1B [chromosome 6], BENC [chromosome 7], and CDK6 [chromosome 7]) upstream or downstream of the BCL11B locus in a process called enhancer hijacking.
- Finally, a translocation cannot be identified in about 20% of cases with deregulated BCL11B overexpression. In such cases, amplification of a downstream enhancer, BCL11B enhancer tandem amplification (BETA), leads to BCL11B promoter driven transcription.
- There is a high prevalence of FLT3 alterations and JAK/STAT activation in acute leukemias driven by genomic alterations leading to BCL11B overexpression.
- RAS and JAK-STAT pathway variants were common in the T/M MPAL and ETP ALL cases, while the PI3K signaling pathway is more commonly altered in T-ALL. For T/M MPAL, the most commonly altered signaling pathway gene was FLT3 (43% of cases). FLT3 variants tended to be mutually exclusive with RAS pathway variants.
- Genes encoding epigenetic regulators (e.g., EZH2 and PHF6) were altered in approximately two-thirds of T/M MPAL cases.
Gene polymorphisms in drug metabolic pathways
Several polymorphisms of genes involved in the metabolism of chemotherapeutic agents have been reported to have prognostic significance in childhood ALL.
- TPMT.
Patients with variant phenotypes of TPMT (a gene involved in the metabolism of thiopurines such as mercaptopurine) appear to have more favorable outcomes, although such patients may also be at higher risk of developing significant treatment-related toxicities, including myelosuppression, infection, and second malignancies. Patients with homozygosity for TPMT variants associated with low enzymatic activity tolerate only very low doses of mercaptopurine (approximately 10% of the standard dose) and are treated with reduced doses of mercaptopurine to avoid excessive toxicity. Patients who are heterozygous for this variant enzyme gene generally tolerate mercaptopurine without serious toxicity, but they do require more frequent dose reductions for hematologic toxicity than do patients who are homozygous for the normal allele.
- NUDT15.
Germline variants in NUDT15 that reduce or abolish activity of this enzyme also lead to diminished tolerance to thiopurines. The NUDT15 variants are most common in East Asian and Hispanic patients, and they are rare in European and African patients. Patients homozygous for the risk variants tolerate only very low doses of mercaptopurine, while patients heterozygous for the risk alleles tolerate lower doses than do patients homozygous for the wild-type allele (approximately 25% dose reduction on average), but there is broad overlap in tolerated doses between the two groups.
- CEP72.
Gene polymorphisms may also affect the expression of proteins that play central roles in the cellular effects of anticancer drugs. As an example, patients who are homozygous for a polymorphism in the promoter region of CEP72 (a centrosomal protein involved in microtubule formation) are at increased risk of vincristine neurotoxicity.
- Single nucleotide polymorphisms.
Genome-wide polymorphism analysis has identified specific single nucleotide polymorphisms associated with high end-induction MRD and risk of relapse. Polymorphisms of interleukin-15, as well as genes associated with the metabolism of etoposide and methotrexate, were significantly associated with treatment response in two large cohorts of ALL patients treated on SJCRH and COG protocols. Polymorphic variants involving the reduced folate carrier and methotrexate metabolism have been linked to toxicity and outcome. While these associations suggest that individual variations in drug metabolism can affect outcome, few studies have attempted to adjust for these variations. It is unknown whether individualized dose modification on the basis of these findings will improve outcomes.
For information about the treatment of childhood ALL, see Childhood Acute Lymphoblastic Leukemia Treatment.
Acute Myeloid Leukemia (AML)
Cytogenetic/molecular features of AML
Genetic analysis of leukemia blast cells (using both conventional cytogenetic methods and molecular methods) is performed on children with AML because both chromosomal and molecular abnormalities are important diagnostic and prognostic markers. Clonal chromosomal abnormalities are identified in the blasts of about 75% of children with AML and are useful in defining subtypes with both prognostic and therapeutic significance. Detection of molecular abnormalities can also aid in risk stratification and treatment allocation. For example, variants of NPM and CEBPA are associated with favorable outcomes, while certain variants of FLT3 portend a high risk of relapse. Identifying the latter variants may allow for targeted therapy.
Comprehensive molecular profiling of pediatric and adult AML has shown that AML is a disease demonstrating both commonalities and differences across the age spectrum.
- Pediatric AML, in contrast to AML in adults, is typically a disease of recurring chromosomal alterations. For a list of common gene fusions and other recurring genomic alterations, see Table 3. Within the pediatric age range, certain gene fusions occur primarily in children younger than 5 years (e.g., NUP98, KMT2A, and CBFA2T3::GLIS2 gene fusions), while others occur primarily in children aged 5 years and older (e.g., RUNX1::RUNX1T1, CBFB::MYH11, and PML::RARA gene fusions).
- In general, pediatric patients with AML have low rates of variants. Most cases show less than one somatic change in protein-coding regions per megabase. This variant rate is somewhat lower than that observed in adult AML and is much lower than the variant rate for cancers that respond to checkpoint inhibitors (e.g., melanoma).
- The pattern of gene variants differs between pediatric and adult AML cases. For example, IDH1, IDH2, TP53, RUNX1, and DNMT3A variants are more common in adult AML than in pediatric AML, while NRAS and WT1 variants are significantly more common in pediatric AML.
- The genomic landscape of pediatric AML cases can change from diagnosis to relapse, with variants detectable at diagnosis dropping out at relapse and, conversely, with new variants appearing at relapse. In a study of 20 cases for which sequencing data were available at diagnosis and relapse, a key finding was that the variant allele frequency at diagnosis strongly correlated with persistence of variants at relapse. Approximately 90% of the diagnostic variants with variant allele frequency greater than 0.4 persisted to relapse, compared with only 28% with variant allele frequency less than 0.2 (P< .001). This observation is consistent with previous results showing that presence of a variant in the FLT3 gene resulting from internal tandem duplications (ITD) predicted for poor prognosis only when there was a high FLT3 ITD allelic ratio.
The 5th edition (2022) of the World Health Organization (WHO) Classification of Hematolymphoid Tumors, as well as the Inaugural WHO Classification of Pediatric Tumors, emphasize a multilayered approach to AML classification. These classifications consider multiple clinico-pathological parameters and seek a genetic basis for disease classification wherever possible. These karyotypic abnormalities and other genomic alterations are used to define specific pediatric AML entities and are outlined in Table 3.
In addition to the cytogenetic/molecular abnormalities that aid AML diagnosis, as defined by the WHO, there are additional entities that, while not disease-defining, have prognostic significance in pediatric AML. All prognostic abnormalities, both those defined by the WHO and these additional abnormalities, have been clustered according to favorable or unfavorable prognosis, as defined by contemporary Children's Oncology Group (COG) clinical trials. These entities are summarized below. After these entities are described, information about additional cytogenetic/molecular and phenotypic features associated with pediatric AML will be described. However, these additional features may not, at present, be used to aid in risk stratification and treatment.
While the t(15;17) fusion that results in the PML::RARA gene product is defined as a pediatric AML risk-defining lesion, given its association with acute promyelocytic leukemia, it is discussed in Childhood Acute Promyelocytic Leukemia.
| Diagnostic Category | Approximate Prevalence in Pediatric AML |
|---|---|
| aAdapted from Pfister et al. | |
| bCryptic chromosomal translocation. | |
| AML with t(8;21)(q22;q22); RUNX1::RUNX1T1 | 13%–14% |
| AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB::MYH11 | 4%–9% |
| APL with t(15;17)(q24.1;q21.2); PML::RARA | 6%–11% |
| AML with KMT2A rearrangement | 25% |
| AML with t(6;9)(p23;q34.1); DEK::NUP214 | 1.7% |
| AML with inv(3)(q21q26)/t(3;3)(q21;q26); GATA2, RPN1::MECOM | <1% |
| AML with ETV6 fusion | 0.8% |
| AML with t(8;16)(p11.2;p13.3); KAT6A::CREBBP | 0.5% |
| AML with t(1;22)(p13.3;q13.1); RBM15::MRTFA (MKL1) | 0.8% |
| AML with CBFA2T3::GLIS2 (inv(16)(p13q24))b | 3% |
| AML with NUP98 fusionb | 10% |
| AML with t(16;21)(p11;q22); FUS::ERG | 0.3%–0.5% |
| AML with NPM1 variant | 8% |
| AML with variants in the bZIP domain of CEBPA | 5% |
Specific recurring cytogenetic and molecular abnormalities are briefly described below. The abnormalities are listed by those in clinical use that identify patients with favorable or unfavorable prognosis, followed by other abnormalities. The nomenclature of the 5th edition of the WHO classification is incorporated for disease entities where relevant.
Abnormalities associated with a favorable prognosis
Cytogenetic/molecular abnormalities associated with a favorable prognosis include the following:
- Core-binding factor (CBF) AML includes cases with RUNX1::RUNX1T1 and CBFB::MYH11 gene fusions.
- AML with RUNX1::RUNX1T1 gene fusions (t(8;21)(q22;q22.1)). In leukemias with t(8;21), the RUNX1 gene on chromosome 21 is fused with the RUNX1T1 gene on chromosome 8. The t(8;21) translocation is associated with the FAB M2 subtype and with granulocytic sarcomas. Adults with t(8;21) have a more favorable prognosis than do adults with other types of AML. The t(8;21) translocation occurs in approximately 12% of children with AML and is associated with a more favorable outcome than AML characterized by normal or complex karyotypes. Overall, the translocation is associated with 5-year overall survival (OS) rates of 74% to 90%.
- AML with CBFB::MYH11 gene fusions (inv(16)(p13.1;q22) or t(16;16)(p13.1;q22)). In leukemias with inv(16), the CBFB gene at chromosome band 16q22 is fused with the MYH11 gene at chromosome band 16p13. The inv(16) translocation is associated with the FAB M4Eo subtype and confers a favorable prognosis for both adults and children with AML. Inv(16) occurs in 7% to 9% of children with AML, for whom the 5-year OS rate is approximately 85%.
Cases with CBFB::MYH11 or RUNX1::RUNX1T1 fusions have distinctive secondary variants, with CBFB::MYH11 secondary variants primarily restricted to genes that activate receptor tyrosine kinase signaling (NRAS, FLT3, and KIT). The prognostic significance of activating KIT variants in adults with CBF AML has been studied with conflicting results. A meta-analysis found that KIT variants appear to increase the risk of relapse without an impact on OS for adults with AML and RUNX1::RUNX1T1 fusions. The prognostic significance of KIT variants in pediatric CBF AML remains unclear. Some studies have found no impact of KIT variants on outcomes, although, in some instances, the treatment used was heterogenous, potentially confounding the analysis. Other studies have reported a higher risk of treatment failure when KIT variants are present. An analysis of a subset of pediatric patients treated with a uniform chemotherapy backbone on the COG AAML0531 study demonstrated that the subset of patients with KIT exon 17 variants had inferior outcomes, compared with patients with CBF AML who did not have the variant. However, treatment with gemtuzumab ozogamicin abrogated this negative prognostic impact. While there was a trend toward inferior outcomes for patients with CBF AML with co-occurring KIT exon 8 abnormalities, this finding was not statistically significant. A second study of 46 patients who were treated uniformly found that KIT exon 17 variants only had prognostic significance in AML with RUNX1::RUNX1T1 fusions but not CBFB::MYH11 fusions.
While KIT variants are seen in both CBF AML subsets, other secondary variants tend to cluster with one of the two fusions. For example, patients with RUNX1::RUNX1T1 fusions also have frequent variants in genes regulating chromatin conformation (e.g., ASXL1 and ASXL2) (40% of cases) and genes encoding members of the cohesin complex (20% of cases). Variants in ASXL1 and ASXL2 and variants in members of the cohesin complex are rare in cases with leukemia and CBFB::MYH11 fusions. Despite this correlation, a study of 204 adults with AML and RUNX1::RUNX1T1 fusions found that ASXL2 variants (present in 17% of cases) and ASXL1 or ASXL2 variants (present in 25% of cases) lacked prognostic significance. Similar results, albeit with smaller numbers, were reported for children with the same abnormalities.
- AML with NPM1 variant. NPM1 is a protein that has been linked to ribosomal protein assembly and transport, as well as being a molecular chaperone involved in preventing protein aggregation in the nucleolus. Immunohistochemical methods can be used to accurately identify patients with NPM1 variants by the demonstration of cytoplasmic localization of NPM. Variants in the NPM1 protein that diminish its nuclear localization are primarily associated with a subset of AML with a normal karyotype, absence of CD34 expression, and an improved prognosis in the absence of FLT3 ITD variants in adults and younger adults.
Studies of children with AML suggest a lower rate of occurrence of NPM1 variants in children compared with adults with normal cytogenetics. NPM1 variants occur in approximately 8% of pediatric patients with AML and are uncommon in children younger than 2 years. NPM1 variants are associated with a favorable prognosis in patients with AML characterized by a normal karyotype. For the pediatric population, conflicting reports have been published regarding the prognostic significance of an NPM1 variant when a FLT3 ITD variant is also present. One study reported that an NPM1 variant did not completely abrogate the poor prognosis associated with having a FLT3 ITD variant, but other studies showed no impact of a FLT3 ITD variant on the favorable prognosis associated with an NPM1 variant.
- AML with CEBPA variants. Variants in the CEBPA gene occur in a subset of children and adults with cytogenetically normal AML. In adults younger than 60 years, approximately 15% of cytogenetically normal AML cases have variants in CEBPA. Outcomes for adults with AML with CEBPA variants appear to be relatively favorable and similar to that of patients with CBF leukemias. Initial studies in adults with AML demonstrated that CEBPA double-variant, but not single-variant, abnormalities were independently associated with a favorable prognosis, leading to the WHO 2016 revision that required biallelic variants for the disease definition. However, a study of over 4,700 adults with AML found that patients with single CEBPA variants in the bZIP C-terminal domain have clinical characteristics and favorable outcomes similar to those of patients with double-variant CEBPA AML.
CEBPA variants occur in approximately 5% of children with AML and have been preferentially found in the cytogenetically normal subtype of AML with FAB M1 or M2.
- Patients with double CEBPA variants or with single CEBPA bZIP variants have a median age of presentation of 12 to 13 years and have gene expression profiles that are highly related to each other.
- Approximately 80% of pediatric patients have double-variant alleles (i.e., cases with both a CEBPA TAD domain and a CEBPA bZIP domain variant), which is predictive of significantly improved survival, similar to the effect observed in adult studies.
- In a study of nearly 3,000 children with AML, both patients with CEBPA double variants and those with only a bZIP domain variant were observed to have a favorable prognosis, compared with patients with wild-type CEBPA.
Given these findings in pediatric AML with CEBPA variants, the presence of a bZIP variant alone confers a favorable prognosis. Importantly, however, there is a small subset of patients with AML and CEBPA variants who have less-favorable outcomes. Specifically, CSF3R variants occur in 10% to 15% of patients with AML and CEBPA variants. CSF3R variants appear to be associated with an increased risk of relapse, but without an impact on OS. At present, the occurrence of this secondary variant does not result in stratification to more intensified therapy in pediatric patients with AML.
While not common, a small percentage of children with AML and CEBPA variants may have an underlying germline variant. In newly diagnosed patients with double-variant CEBPA AML, germline screening should be considered in addition to usual family history queries because 5% to 10% of these patients have a germline CEBPA abnormality that confers an increased malignancy risk.
Cytogenetic abnormality associated with a variable prognosis: KMT2A (MLL) gene rearrangements
The 5th edition (2022) of the WHO Classification of Hematolymphoid Tumors includes a diagnostic category of AML with KMT2A rearrangements. Specific translocation partners are not listed because there are more than 80 KMT2A fusion partners.
- KMT2A gene rearrangements occur in approximately 20% of children with AML. These cases, including most AMLs secondary to epipodophyllotoxin exposure, are generally associated with monocytic differentiation (FAB M4 and M5). KMT2A rearrangements are also reported in approximately 10% of FAB M7 (AMKL) patients.
- The median age for 11q23/KMT2A-rearranged cases in children is approximately 2 years, and most translocation subgroups have a median age at presentation of younger than 5 years. However, significantly older median ages are seen at presentation of pediatric cases with t(6;11)(q27;q23) (12 years) and t(11;17)(q23;q21) (9 years).
- Outcomes for patients with de novo AML and KMT2A gene rearrangements are generally similar to or slightly worse than the outcomes observed in other patients with AML. As the KMT2A gene can participate in translocations with many different fusion partners, the specific fusion partner appears to influence prognosis. This finding was demonstrated by a large international retrospective study that evaluated the outcomes of 756 children with 11q23- or KMT2A-rearranged AML and a second COG analysis that studied KMT2A outcomes within the context of the AAML0531 trial.
- The most common translocation, representing approximately 50% of KMT2A-rearranged cases in the pediatric AML population, is t(9;11)(p22;q23), in which the KMT2A gene is fused with the MLLT3 gene. Single clinical trial groups have variably described a more favorable prognosis for these patients. However, neither the international retrospective study nor the COG study confirmed the favorable prognosis for this subgroup. This fusion, which is associated with an intermediate prognosis, is not currently classified as high risk, at least within the COG, unless minimal residual disease (MRD) remains at the end of induction 1.
- KMT2A-rearranged AML subgroups that are associated with poor outcomes include the following:
- Cases with the t(10;11) translocation are a group at high risk of relapse in bone marrow and the CNS. Some cases with the t(10;11) translocation have fusion of the KMT2A gene with the MLLT10 gene at 10p12, while others have fusion of KMT2A with ABI1 at 10p11.2. An international retrospective study found that these cases, which present at a median age of approximately 1 to 3 years, have a 5-year event-free survival (EFS) rate of 17% to 30%.
- Patients with t(6;11)(q27;q23) (KMT2A::AFDN) have poor outcomes, with 5-year EFS rates of 11% to 15%.
- Patients with t(4;11)(q21;q23) (KMT2A::AFF1) often present with hyperleukocytosis and also have poor outcomes, with 5-year EFS rates of 0% to 29%.
- Patients with t(11;19)(q23;p13.3) (KMT2A::MLLT1) have poor outcomes, with a 5-year EFS rate of 14%.
- Based on the data above, the International Berlin-Frankfurt-Münster (iBFM) study group analyzed data regarding outcomes in patients with KMT2A rearrangements enrolled in BFM, COG, and other European cooperative group studies. In keeping with earlier papers, this study classified patients with 6q27 (KMT2A::AFDN, i.e., MLLT4), 4q21 (KMT2A::AFF1, i.e., MLL::MLLT2), 10p12.3 (KMT2A::MLLT10), 10p12.1 (KMT2A::ABI1), and 19p13.3 (KMT2A::MLLT1, i.e., MLL::ENL) as high risk, while all others were considered in the non–high-risk group. Using this classification, the 5-year EFS rates for patients with non–high-risk, KMT2A-rearranged AML is 54%, compared with 30.3% for patients with high-risk disease. MRD assessment after induction 2 imparts further prognostic significance within the iBFM analysis. However, MRD at the end of induction 1 did not predict for relapse in the context of the COG analysis.
- With this distinction, non–high-risk KMT2A fusions are, in most cooperative groups, upstaged to high-risk if MRD is noted after induction treatment.
- When examining outcomes for patients with KMT2A rearrangements, both overall and within the context of high-risk and non–high-risk fusions, treatment with gemtuzumab ozogamicin appeared to abrogate the negative prognostic impact of the variant. Specifically, the EFS rate for patients with KMT2A-rearranged AML was superior with gemtuzumab ozogamicin treatment than without this treatment (48% vs. 29%; P = .003) and comparable with the outcomes observed in patients without KMT2A rearrangements.
Cytogenetic/molecular abnormalities associated with an unfavorable prognosis
Genetic abnormalities associated with an unfavorable prognosis are described below. Some of these are disease-defining alterations that are initiating events and maintained throughout a patient's disease course. Other entities described below are secondary alterations (e.g., FLT3 alterations). Although these secondary alterations do not induce disease on their own, they are able to promote the cell growth and survival of leukemias that are driven by primary genetic alterations.
- AML with GATA2 or MECOM abnormalities (inv(3)(q21.3;q26.2)/t(3;3)(q21.3;q26.2) or t(3;21)(26.2;q22)). MECOM at chromosome 3q26 codes for two proteins, EVI1 and MDS1::EVI1, both of which are transcription regulators. The inv(3) and t(3;3) abnormalities lead to overexpression of EVI1 and to reduced expression of GATA2. These abnormalities are associated with poor prognosis in adults with AML but are rare in children (<1% of pediatric AML cases).
Abnormalities involving MECOM can also be detected in some AML cases with other 3q abnormalities (e.g., t(3;21)(26.2;q22)). The RUNX1::MECOM fusion is also associated with poor prognosis.
- AML with NPM1::MLF1 (t(3;5)(q25;q34)) gene fusions. This fusion results in a chimeric protein that includes virtually the entire MLF1 gene. This gene does not usually have a function in normal hematopoiesis, but in this context, it is hypothesized to result in ectopic expression of the protein. While incredibly rare in pediatrics (less than 0.5% of cases, most of which occur in adolescence), it is generally associated with poor prognosis.
- AML with DEK::NUP214 (t(6;9)(p23;q34.1)) gene fusions. t(6;9) leads to the formation of a leukemia-associated fusion protein DEK::NUP214. This subgroup of AML has been associated with a poor prognosis in adults with AML, and occurs infrequently in children (less than 1% of AML
cases). The median age of children with AML and DEK::NUP214 fusions is 10 to 11 years, and approximately 40% of pediatric patients have FLT3 ITD.
t(6;9) AML appears to be associated with a high risk of treatment failure in children, particularly for those not proceeding to allogeneic HSCT.
- AML with KAT6A::CREBBP (t(8;16)(p11.2;p13.3)) gene fusions (if 90 days or older at diagnosis). The t(8;16) translocation fuses the KAT6A gene on chromosome 8p11 to CREBBP on chromosome 16p13. It is associated with poor outcomes in adults, although its prognostic significance in pediatrics is less clear. Although this translocation rarely occurs in children, in an iBFM AML study of 62 children, this translocation was associated with younger age at diagnosis (median, 1.2 years), FAB M4/M5 phenotype, erythrophagocytosis, leukemia cutis, and disseminated intravascular coagulation. A substantial proportion of infants diagnosed with t(8;16) AML in the first month of life show spontaneous remission, although AML recurrence may occur months to years later. These observations suggest that a watch-and-wait approach could be considered in cases of t(8;16) AML diagnosed in the neonatal period if close long-term monitoring can be ensured. For older children, the prognosis is less favorable, and the typical recommendation is to proceed to HSCT once remission is achieved.
- AML with FUS::ERG (t(16;21)(p11;q22)) gene fusions. In leukemias with t(16;21)(p11;q22), the FUS gene is joined with the ERG gene, producing a distinctive AML subtype with a gene expression profile that clusters separately from other cytogenetic subgroups. This fusion is rare in pediatrics and represents 0.3% to 0.5% of pediatric AML cases. In a cohort of 31 patients with AML and FUS::ERG fusions, outcomes were poor, with a 4-year EFS rate of 7% and a cumulative incidence of relapse rate of 74%.
- AML with CBFA2T3::GLIS2 gene fusions.CBFA2T3::GLIS2 is a fusion resulting from a cryptic chromosome 16 inversion (inv(16)(p13.3;q24.3)). It occurs commonly in non–Down syndrome AMKL, representing 16% to 27% of pediatric AMKL and presents at a median age of 1 year. Leukemia cells with CBFA2T3::GLIS2 fusions have a distinctive immunophenotype (initially reported as the RAM phenotype), with high CD56, dim or negative expression of CD45 and CD38, and a lack of HLA-DR expression. This fusion is a very high-risk lesion associated with poor clinical outcomes.
In a study of approximately 2,000 children with AML, the CBFA2T3::GLIS2 fusion was identified in 39 cases (1.9%), with a median age at presentation of 1.5 years. All cases observed in children were younger than 3 years. Approximately one-half of cases had M7 megakaryoblastic morphology, and 29% of patients were Black or African American (exceeding the 12.8% frequency in patients lacking the fusion). Children with the fusion were found to be MRD positive after induction 1 in 80% of cases. In an analysis of outcomes from serial COG trials of 37 identified patients, OS at 5 years from study entry was 22.0% for patients with CBFA2T3::GLIS2 fusions versus 63.0% for fusion-negative patients (n = 1,724). Even worse outcomes were demonstrated when the subset of patients with CBFA2T3::GLIS2 AMKL were compared with patients with AMKL without the abnormality. Analysis from the COG AAML0531 and AAML1031 trials revealed OS rates of 43% (± 37%) and 10% (± 19%), respectively, among children with AMKL and this fusion. As CBFA2T3::GLIS2 leukemias express high levels of cell surface FOLR1, a targetable surface antigen by immunotherapeutic approaches, the roles of such agents are planned for study in this high-risk population.
- AML with NUP98 gene fusions.NUP98 has been reported to form leukemogenic gene fusions with more than 20 different partners. A significant proportion of cases are associated with non–Down syndrome AMKL, although approximately 50% are seen outside of that morphologic subtype. The two most common gene fusions in pediatric AML are NUP98::NSD1 and NUP98::KDM5A. In one report, the former fusion was observed in approximately 15% of cytogenetically normal pediatric AML cases, and the latter fusion was observed in approximately 10% of pediatric AMKL cases (see below). AML cases with either NUP98 gene fusion show high expression of HOXA and HOXB genes, indicative of a stem cell phenotype. Some of the less common fusions entail HOX genes.
The NUP98::NSD1 gene fusion, which is often cytogenetically cryptic, results from the fusion of NUP98 (chromosome 11p15) with NSD1 (chromosome 5q35). This alteration occurs in approximately 4% to 7% of pediatric AML cases. It is the most common NUP98 fusion seen. This disease phenotype is characterized by the following:
- The highest frequency of NUP98::NSD1 fusions in the pediatric population is observed in children aged 5 to 9 years (approximately 8%), with a lower frequency in younger children (approximately 2% in children younger than 2 years).
- Patients with NUP98::NSD1 fusions present with a high white blood cell (WBC) count (median, 147 × 109/L in one study). Most patients with AML and NUP98::NSD1 fusions do not show cytogenetic aberrations. There is a slight male predominance for patients with this fusion (64.5% vs. 32.2%).
- A high percentage of patients with NUP98::NSD1 fusions (74%–90%) have co-occurring FLT3 ITD AML.
- In one of a series of COG studies, 108 children with NUP98::NSD1 fusions demonstrated lower rates of complete remission (CR) (38%, P< .001) and higher rates of MRD (73%, P< .001), compared with a cohort of patients without NUP98 fusions. Patients with NUP98::NSD1 fusions also had inferior EFS rates (17% vs. 47%; P< .001) and OS rates (36% vs. 64%; P< .001), compared with the reference cohort. In another study that included children (n = 38) and adults (n = 7) with AML and NUP98::NSD1 fusions, presence of both NUP98::NSD1 fusions and FLT3 ITD independently predicted poor prognosis. Patients with both lesions had a low CR rate (approximately 30%) and a low 3-year EFS rate (approximately 15%).
- In a study of children with refractory AML, NUP98 was overrepresented compared with a cohort who did achieve remission (21% [6 of 28 patients] vs. <4%).
A cytogenetically cryptic translocation, t(11;12)(p15;p13), results in the NUP98::KDM5A gene fusion. Approximately 2% of all pediatric AML patients have NUP98::KDM5A fusions, and these cases tend to present at a young age (median age, 3 years). Additional clinical characteristics are as follows:
- Cases with NUP98::KDM5A fusions tend to be AMKL (34%), followed by FAB M5 (21%), and FAB M6 (17%) histologies. NUP98::KDM5A fusions are observed in approximately 10% of pediatric AMKL cases, and patients with this fusion tend to present with lower WBC counts than patients with NUP98::NSD1 fusions.
- Other genetic aberrations associated with pediatric AML, including FLT3 variants, are uncommon in patients with NUP98::KDM5A fusions.
- Prognosis for children with NUP98::KDM5A fusions is inferior to that of other children with AML (5-year EFS rate, 29.6% ± 14.6%; OS rate, 34.1% ± 16.1%) in one series. Another study that included 32 patients with NUP98::KDM5A fusions demonstrated similar CR rates to the reference population but inferior OS (30%, P< .001) and EFS rates (25%; P = .01).
- AML with 12p13.2 rearrangements (ETV6 and any partner gene). The ETS family of genes encode transcription factors responsible for cellular growth and development. The ETV6 gene encodes a transcription factor that serves as a tumor suppressor gene and is the most frequent ETS family rearranged partner in pediatric AML. The cryptic translocation t(7;12)(q36;p13) encodes ETV6::MNX1, the most frequent ETV6-rearranged fusion partner, which occurs in approximately 1% of pediatric AML cases (enriched in infants). It is associated with poor clinical outcomes. It is also strongly associated with trisomy 19. The transcription may be cryptic by conventional karyotyping and, in some cases, may be confirmed only by fluorescence in situ hybridization (FISH). This alteration occurs virtually exclusively in children younger than 2 years, with a median age of diagnosis of 6 months. It appears to be associated with a high risk of treatment failure. A literature review of 17 cases showed a 3-year EFS rate of 24% and OS rate of 42%.
- AML with 12p deletion to include 12p13.2 (loss of ETV6).ETV6 deletions are exceedingly rare in pediatric AML. In one pediatric series, 4 of 259 patients (1.5%) had an ETV6 deletion. This abnormality is enriched in adult patients with chromosome 7 abnormalities and in patients with TP53 variants. However, in a second pediatric series, there was a reported correlation with CBF AML. According to this latter series, relapse risk rates for patients with and without deletions in ETV6 were 63% and 45%, respectively (P = .3), with corresponding disease-free survival (DFS) rates of 32% and 53%, respectively (P = .2). However, there was a high prevalence of CBF AML in patients with ETV6 deletions. In the context of CBF AML, the deletion was associated with adverse outcomes. Patients with CBF AML, with and without ETV6 deletions, had EFS rates of 0% and 63%, respectively (P = .002). Of the patients with CBF AML who achieved an initial CR, those with an ETV6 deletion had a risk of relapse rate of 88%, compared with 38% for those without the deletion (P = .08). The corresponding DFS rates were 0% for patients with an ETV6 deletion, compared with 61% for those without the deletion (P = .009).
- Chromosome 5 and 7 abnormalities. Chromosomal abnormalities associated with poor prognosis in adults with AML include those involving chromosome 5 (del(5q)) and chromosome 7 (monosomy 7). These cytogenetic subgroups represent approximately 2% and 4% of pediatric AML cases, respectively, and are also associated with poor prognosis in children. Chromosome 5 and 7 abnormalities appear to lack prognostic significance in AML patients with Down syndrome who are aged 4 years and younger.
In the past, patients with del(7q) were also considered to be at high risk of treatment failure, and data from adults with AML support a poor prognosis for both del(7q) and monosomy 7. However, outcome for children with del(7q), but not monosomy 7, appears comparable to that of other children with AML. The presence of del(7q) does not abrogate the prognostic significance of favorable cytogenetic characteristics (e.g., inv(16) and t(8;21)).
- AML with 10p12.3 rearrangements (MLLT10::any partner gene).MLLT10 frequently forms fusions with partners other than KMT2A, and these fusions are also associated with a poor prognosis. A retrospective review of 2,226 children enrolled in serial COG trials identified 23 children with non-KMT2A::MLLT10 fusions. Nearly one-half of patients (13 of 23) had MLLT10::PICALM fusions, and the EFS rate of this heterogenous group was 12.7%. Another study focused on the prognostic impact of the MLLT10::PICALM fusion, which results in aberrant hematopoiesis and loss of chromatin-mediated gene regulation. Within this specific subset, the 20 pediatric patients with MLLT10::PICALM fusions had a poor prognosis. The 5-year EFS rate was 22%, and the OS rate was 26%.
- FLT3 variants. Presence of a FLT3 ITD variant appears to be associated with poor prognosis in adults with AML, particularly when both alleles are altered or there is a high ratio of the variant allele to the normal allele. FLT3 ITD variants also convey a poor prognosis in children with AML. The frequency of FLT3 ITD variants in children is lower than that observed in adults, especially for children younger than 10 years, for whom 5% to 10% of cases have the variant (compared with approximately 30% in adults).
The prevalence of FLT3 ITD is increased in certain genomic subtypes of pediatric AML, including cases with the NUP98::NSD1 gene fusion, 80% to 90% of which have a co-occurring FLT3 ITD.
The prognostic significance of FLT3 ITD is modified by the presence of other recurring genomic alterations. For patients who have FLT3 ITD, the presence of either WT1 variants or NUP98::NSD1 fusions is associated with poorer outcomes (EFS rates below 25%) than for patients who have FLT3 ITD without these alterations. Conversely, a co-occurring cryptic DEK::NUP214 fusion may be more favorable, particularly with the addition of a FLT3 inhibitor to standard front-line chemotherapy. When FLT3 ITD is accompanied by NPM1 variants, the outcome is relatively favorable and is similar to that of pediatric AML cases without FLT3 ITD. The latter subset is the one scenario in which the presence of the FLT3 ITD variant does not necessarily upstage a patient to high risk, based on the favorable outcomes seen with the co-occurring variants.
Activating point variants of FLT3 have also been identified in both adults and children with AML, although the clinical significance of these variants is not clearly defined. Some of these point variants appear to be specific to pediatric patients.
- RAM phenotype. The RAM phenotype is characterized by high-intensity CD56 expression, dim-to-negative expression of CD45 and CD38, and a lack of HLA-DR expression. These patients tend to be younger, with a median age of 1.6 years in the initially reported series. This phenotype is enriched in patients with non-Down syndrome–related AMKL. Clinically, patients with the RAM phenotype have inferior outcomes. In the initial series, patients in the RAM cohort had a 3-year EFS rate of 16%, compared with 51% for patients in the non-RAM cohort (P< .001). Patients in the RAM cohort also had inferior survival compared with patients with high CD56 expression, who lacked other phenotypic features of the RAM phenotype. OS was also inferior compared with the patients without the RAM phenotype (26% vs. 69%, P = .001). In a subanalysis, the OS of the patients in the RAM cohort was also markedly worse than patients in the CD56-positive (non-RAM) cohort (26% vs. 66%, P< .001) and the CD56-negative cohort (26% vs. 70%, P< .001). Many, but not all, patients with a RAM phenotype have evidence of a CBFA2T3::GLIS2 fusion that, in itself, confers very high-risk disease. In a published series, approximately 60% of patients with the RAM phenotype at diagnosis were subsequently found to have this cryptic fusion that also confers higher-risk disease.
Additional cytogenetic/molecular abnormalities that may have prognostic significance
This section includes cytogenetic/molecular abnormalities that are seen at diagnosis and do not impact disease risk stratification but may have prognostic significance.
- AML with RUNX1::CBFA2T3 (t(16;21)(q24;q22)) gene fusions. In leukemias with t(16;21)(q24;q22), the RUNX1 gene is fused with the CBFA2T3 gene, and the gene expression profile is closely related to that of AML cases with t(8;21) and RUNX1::RUNX1T1 fusions. Patients present at a median age of 7 years. This cancer is rare, representing approximately 0.1% to 0.3% of pediatric AML cases. Among 23 patients with RUNX1::CBFA2T3 fusions, five presented with secondary AML, including two patients who had a primary diagnosis of Ewing sarcoma. Outcomes were favorable for the cohort of 23 patients, with a 4-year EFS rate of 77% and a cumulative incidence of relapse rate of 0%.
- RAS variants. Although variants in RAS have been identified in 20% to 25% of patients with AML, the prognostic significance of these variants has not been clearly shown. Variants in NRAS are more commonly observed than variants in KRAS in pediatric AML cases. RAS variants occur with similar frequency for all Type II alteration subtypes, with the exception of APL, for which RAS variants are seldom observed.
- AML with RBM15::MRTFA gene fusions. The t(1;22)(p13;q13) translocation that produces RBM15::MRTFA fusions (also known as RBM15::MKL1) is uncommon (<1% of pediatric AML) and is restricted to AMKL. Studies have found that t(1;22)(p13;q13) is observed in 10% to 20% of children with AMKL who have evaluable cytogenetics or molecular genetics. Most AMKL cases with t(1;22) occur in infants, with the median age at presentation (4–7 months) being younger than for other children with AMKL. Cases with detectable RBM15::MKL1 fusion transcripts in the absence of t(1;22) have also been reported because these young patients usually have hypoplastic bone marrow.
An international collaborative retrospective study of 51 t(1;22) cases reported that patients with this abnormality had a 5-year EFS rate of 54.5% and an OS rate of 58.2%, similar to the rates for other children with AMKL. In another international retrospective analysis of 153 cases with non–Down syndrome AMKL who had samples available for molecular analysis, the 4-year EFS rate for patients with t(1;22) was 59% and the OS rate was 70%, significantly better than for AMKL patients with other specific genetic abnormalities (CBFA2T3::GUS2 fusions, NUP98::KDM5A fusions, KMT2A rearrangements, monosomy 7). Similar outcomes were seen in the COG AAML0531 and AAML1031 phase III trials (5-year OS rates, 86% ± 26% [n = 7] and 54% ± 14% [n = 14] for AAML0531 and AAML1031, respectively).
- HOX rearrangements. Cases with a gene fusion involving a HOX cluster gene represented 15% of pediatric AMKL in one report. This report observed that these patients appear to have a relatively favorable prognosis, although the small number of cases studied limits confidence in this assessment.
- GATA1 variants.GATA1-truncating variants in non–Down syndrome AMKL arise in young children (median age, 1–2 years) and are associated with amplification of the RCAN1 gene on chromosome 21. These patients represented approximately 10% of non–Down syndrome AMKL and appeared to have a favorable outcome if there were no prognostically unfavorable fusion genes also present, although the number of patients studied was small (n = 8).
- Hypodiploidy. Hypodiploidy is defined as a modal chromosome number of less than or equal to 45. This occurs rarely in pediatric patients with AML. In a retrospective cohort analysis, the iBFM AML study group aimed to characterize hypodiploidy in pediatric patients with AML. The study excluded several patient groups, including patients with APL, Down syndrome, or loss of chromosome 7. Their observations included the following:
- Hypodiploidy was observed in 1.3% of children with AML. Approximately 80% of patients had a modal chromosome number of 45, and the remaining 20% of patients had a modal chromosome number of either 43 or 44.
- Most patients (>80%) with a modal chromosome number of 43 or 44 also met the criteria for complex karyotype. In this study, a complex karyotype was defined as at least three independent chromosomal abnormalities, regardless of whether these were structural abnormalities or defects in chromosome number, and an absence of recurrent aberrations as defined by the WHO.
- Patients with a modal chromosome number of 43 or 44 had decreased EFS rates and OS rates when compared with patients who had 45 chromosomes (EFS rate, 21% vs. 37%; P = .07; OS rate, 33% vs. 56%; P = .1).
- UBTF tandem duplication.UBTF is located at chromosome 17q21.31, and it codes for a nucleolar protein that interacts with ribosomal DNA to mediate RNA polymerase 1 ribosomal RNA transcription.
- UBTF tandem duplication (UBTF-TD) is mutually exclusive with other leukemia driver genomic alterations. Like other leukemogenic drivers, it is maintained at relapse.
- UBTF genomic alterations involving heterozygous somatic variants resulting in in-frame tandem duplication of UBTF exon 13 are observed in approximately 4% of pediatric AML cases.
- UBTF-TD AML in the pediatric population primarily occurs during adolescence (median age, 12–14 years). It is also observed in adults younger than 60 years, but it is uncommon among AML in older adult patients.
- FLT3 ITD is common in cases of AML with UBTF-TD. Approximately two-thirds of cases have FLT3 ITD. In addition, approximately 40% of cases with UBTF-TD AML have WT1 variants.
- In the AAML1031 clinical trial, EFS and OS rates for patients with UBTF-TD were 30% and 44%, respectively. These values were lower than those for non–UBTF-TD patients enrolled in AAML1031 (45% and 64%, respectively). Outcome for patients with UBTF-TD was similar to that for patients with KMT2A rearrangements.
- In the AAML1031 trial, co-occurrence of UBTF-TD with either FLT3 ITD or WT1 variants was associated with an inferior prognosis, compared with patients with UBTF-TD alone.
- AML with CBFB::GDXY insertions.CBFB encodes the CBFB protein that is part of the multiprotein, core-binding transcription factor complex, which master regulates a gene expression program critical for hematopoiesis. CBFB is recurrently fused with MYH11 in inv(16)/t(16;16) AML.
- In-frame insertions in exon 3 of CBFB have been identified in about 0.4% of pediatric AML cases at diagnosis. All described insertions lead to replacement of aspartic acid at position 87 (D87) with either glycine, aspartic acid, serin, and tyrosine (GDSY) or glycine, aspartic acid, threonine, and tyrosine (GDTY).
- CBFB::GDXY insertions are associated with a gene expression profile overlapping with CBFB::MYH11–expressing AML, with the exception of increased expression of stem cell genes such as HOXA cluster genes and MEIS1.
- CBFB::GDXY insertions frequently co-occur with FLT3 tyrosine kinase domain (TKD) and BCOR1 variants, but lack KIT variants, which are frequently found in CBFB::MYH11 AML.
- CBFB::GDXY insertions appear to be enriched among adolescents and young adults.
- The impact of CBFB::GDXY insertions on patient outcomes are unclear due to a paucity of data. However, early analysis suggests that these patients may not have the same favorable outcome as patients with CBFB::MYH11 fusions.
- RUNX1 variants. AML with RUNX1 variants was a provisional entity in the 2016 WHO classification. In the 5th edition of the WHO classification, it falls into the category of AML with other defined genetic alterations. This subtype of AML is more common in adults than in children. In adults, the RUNX1 variant is associated with a high risk of treatment failure. A meta-analysis of outcomes for adult patients with RUNX1 variants also demonstrated high-risk disease, although this significance was lost in the context of intermediate-risk cytogenetics.
In a study of children with AML, RUNX1 variants were observed in 11 of 503 patients (approximately 2%). Six of 11 patients with AML and RUNX1 variants failed to achieve remission, and their 5-year EFS rate was 9%, suggesting that the RUNX1 variant confers a poor prognosis in both children and adults. However, a second study in which 23 children were found to have RUNX1 variants among 488 children with AML found no significant impact of RUNX1 variants on response or outcome. Additionally, analysis identified that children with RUNX1 variants were more frequently male, adolescents, and had a greater incidence of co-occurring FLT3 ITD and other variants. However, in each of these groups, univariable and multivariable analyses found no survival differences based on the presence of RUNX1 variants. Genetic variants of RUNX1 result in a familial platelet disorder with associated myeloid malignancy (FPD-MM).
- WT1 variants. WT1, a zinc-finger protein regulating gene transcription, is altered in approximately 10% of cytogenetically normal cases of AML in adults. The WT1 variant has been shown in some, but not all, studies to be an independent predictor of worse DFS, EFS, and OS in adult patients.
In children with AML, WT1 variants are observed in approximately 10% of cases. Cases with WT1 variants are enriched among children with normal cytogenetics and FLT3 ITD but are less common among children younger than 3 years. AML cases with NUP98::NSD1 fusions are enriched for both FLT3 ITD and WT1 variants. In univariate analyses, WT1 variants are predictive of poorer outcome in pediatric patients, but the independent prognostic significance of WT1 variant status is unclear because of its strong association with FLT3 ITD and its association with NUP98::NSD1 fusions. The largest study of WT1 variants in children with AML observed that children with WT1 variants in the absence of FLT3 ITD had outcomes similar to that of children without WT1 variants, while children with both WT1 variants and FLT3 ITD had survival rates less than 20%.
In a study of children with refractory AML, WT1 was overrepresented, compared with a cohort who did achieve remission (54% [15 of 28 patients] vs. 15%).
- DNMT3A variants. Variants of the DNMT3A gene have been identified in approximately 20% of adult patients with AML. These variants are uncommon in patients with favorable cytogenetics but occur in one-third of adult patients with intermediate-risk cytogenetics. Variants in this gene are independently associated with poor outcome. DNMT3A variants are virtually absent in children.
- IDH1 and
IDH2 variants. Variants in IDH1 and IDH2, which code for isocitrate dehydrogenase, occur in approximately 20% of adults with AML, and they are enriched in patients with NPM1 variants. The specific variants that occur in IDH1 and IDH2 create a novel enzymatic activity that promotes conversion of alpha-ketoglutarate to 2-hydroxyglutarate. This novel activity appears to induce a DNA hypermethylation phenotype similar to that observed in AML cases with loss-of-function variants in TET2.
Variants in IDH1 and IDH2 are rare in pediatric AML, occurring in 0% to 4% of cases. There is no indication of a negative prognostic effect for IDH1 and IDH2 variants in children with AML.
- CSF3R variants.CSF3R is the gene encoding the granulocyte colony-stimulating factor (G-CSF) receptor, and activating variants in CSF3R are observed in 2% to 3% of pediatric AML cases. These variants lead to enhanced signaling through the G-CSF receptor. They are primarily observed in AML with either CEBPA variants or with CBF abnormalities (RUNX1::RUNX1T1 and CBFB::MYH11 fusions). In a study of 2,150 pediatric patients with AML, 35 patients (1.6%) were found to have CSF3R variants; 30 (89%) of these cases were in patients with either RUNX1::RUNX1T1 fusions (n = 18) or with CEBPA variants (n = 12). Risk of relapse was significantly higher for patients with co-occurring CSF3R and CEBPA variants, compared with patients with RUNX1::RUNX1T1 fusions and CSF3R variants.
Although relapse rates are higher in patients with AML who have co-occurring CSF3R and CEBPA variants, OS is not adversely impacted, reflecting a high salvage rate with reinduction therapy and HSCT.
Activating variants in CSF3R are also observed in patients with severe congenital neutropenia. These variants are not the cause of severe congenital neutropenia, but rather arise as somatic variants and can represent an early step in the pathway to AML. In one study of patients with severe congenital neutropenia, 34% of patients who had not developed a myeloid malignancy had CSF3R variants detectable in peripheral blood neutrophils and mononuclear cells, while 78% of patients who had developed a myeloid malignancy showed CSF3R variants. A study of 31 patients with severe congenital neutropenia who developed AML or MDS observed CSF3R variants in approximately 80% of patients. The study also observed a high frequency of RUNX1 variants (approximately 60%), suggesting cooperation between CSF3R and RUNX1 variants for leukemia development within the context of severe congenital neutropenia.
For information about the treatment of childhood AML, see Childhood Acute Myeloid Leukemia Treatment.
Acute Promyelocytic Leukemia (APL)
RARA Fusion Proteins
The characteristic chromosomal abnormality associated with acute promyelocytic leukemia (APL) is t(15;17)(q22;q21). This translocation involves a breakpoint that includes the retinoic acid receptor and leads to production of the PML::RARA fusion protein. Other more complex chromosomal rearrangements may also lead to a PML::RARA fusion and result in APL.
Patients with a suspected diagnosis of APL can have their diagnosis confirmed by detection of the PML::RARA fusion protein through fluorescence in situ hybridization (FISH), reverse transcriptase–polymerase chain reaction (RT-PCR), or conventional cytogenetics. Quantitative RT-PCR allows identification of the three common transcript variants and is used for monitoring response on treatment and early detection of molecular relapse. In addition, an immunofluorescence method using an anti-PML monoclonal antibody can rapidly establish the presence of the PML::RARA fusion protein based on the characteristic distribution pattern of PML that occurs in the presence of the fusion protein.
Uncommon molecular variants of APL produce fusion proteins that join distinctive gene partners (e.g., PLZF, NPM, STAT5B, and NuMA) to RARA. Recognition of these rare variants is important because they differ in their sensitivities to tretinoin and arsenic trioxide.
- PLZF::RARA fusion gene variant. The PLZF::RARA variant, characterized by t(11;17)(q23;q21), represents about 0.8% of APL, expresses surface CD56, and has very fine granules, compared with t(15;17) APL. APL with the PLZF::RARA fusion gene has been associated with a poor prognosis and usually does not respond to tretinoin or arsenic trioxide.
- NPM::RARA or NuMA::RARA fusion gene variants. The rare APL variants with NPM::RARA (t(5;17)(q35;q21)) or NuMA::RARA (t(11;17)(q13;q21)) translocations may still be responsive to tretinoin.
- PML::RARA fusion gene variant. There are rare case reports of patients with PML::RARA fusion–negative APL. One such APL is the torque teno mini virus (TTMV) subtype. This is a newly described entity in which the TTMV genome is integrated into intron 2 of the human RARA gene, resulting in a TTMV::RARA gene fusion. The clinical and morphological features of this APL subtype are similar to those of PML::RARA fusion–positive APL.
FLT3 Variants
FLT3 variants (either internal tandem duplication or tyrosine kinase domain variants) are observed in 40% to 50% of APL cases. The presence of FLT3 variants is correlated with higher white blood cell counts and the microgranular variant (M3v) subtype. The FLT3 variant has previously been associated with an increased risk of induction death and, in some reports, an increased risk of treatment failure. Given the extremely high cure rates for children with APL who were treated with tretinoin and arsenic trioxide, FLT3 variants are not associated with inferior outcomes.
For information about the treatment of childhood APL, see Childhood Acute Promyelocytic Leukemia Treatment.
Chronic Myeloid Leukemia (CML)
Genomics of CML
The cytogenetic abnormality required for diagnosis of CML is the Philadelphia chromosome (Ph), which represents a translocation of chromosomes 9 and 22 (t(9;22)), resulting in a BCR::ABL1 fusion protein.
Additional chromosomal abnormalities have been found in studies of adults with CML in the TKI era. These studies have illustrated a number of adverse prognostic variants, including those identified as high risk in the chronic phase.
For information about the treatment of childhood CML, see Childhood Chronic Myeloid Leukemia Treatment.
Juvenile Myelomonocytic Leukemia (JMML)
Molecular Features of JMML
The genomic landscape of JMML is characterized by variants in one of five genes of the RAS pathway: NF1, NRAS, KRAS, PTPN11, and CBL. In a series of 118 consecutively diagnosed JMML cases with RAS pathway–activating variants, PTPN11 was the most commonly altered gene, accounting for 51% of cases (19% germline and 32% somatic) (see Figure 5). Patients with NRAS variants accounted for 19% of cases, and patients with KRAS variants accounted for 15% of cases. NF1 variants accounted for 8% of cases, and CBL variants accounted for 11% of cases. Although variants among these five genes are generally mutually exclusive, 4% to 17% of cases have variants in two of these RAS pathway genes, a finding that is associated with poorer prognosis.
The variant rate in JMML leukemia cells is very low, but additional variants beyond those of the five RAS pathway genes described above are observed. Secondary genomic alterations are observed for genes of the transcriptional repressor complex PRC2 (e.g., ASXL1 was altered in 7%–8% of cases). Some genes associated with myeloproliferative neoplasms in adults are also altered at low rates in JMML (e.g., SETBP1 was altered in 6%–9% of cases). JAK3 variants are also observed in a small percentage (4%–12%) of JMML cases. Cases with germline PTPN11 and germline CBL variants showed low rates of additional variants (see Figure 5). The presence of variants beyond disease-defining RAS pathway variants is associated with an inferior prognosis.
A report describing the genomic landscape of JMML found that 16 of 150 patients (11%) lacked canonical RAS pathway variants. Among these 16 patients, 3 were observed to have in-frame fusions involving receptor tyrosine kinases (DCTN1::ALK, RANBP2::ALK, and TBL1XR1::ROS1 gene fusions). These patients all had monosomy 7 and were aged 56 months or older. One patient with an ALK gene fusion was treated with crizotinib plus conventional chemotherapy and achieved a complete molecular remission and proceeded to allogeneic bone marrow transplant.
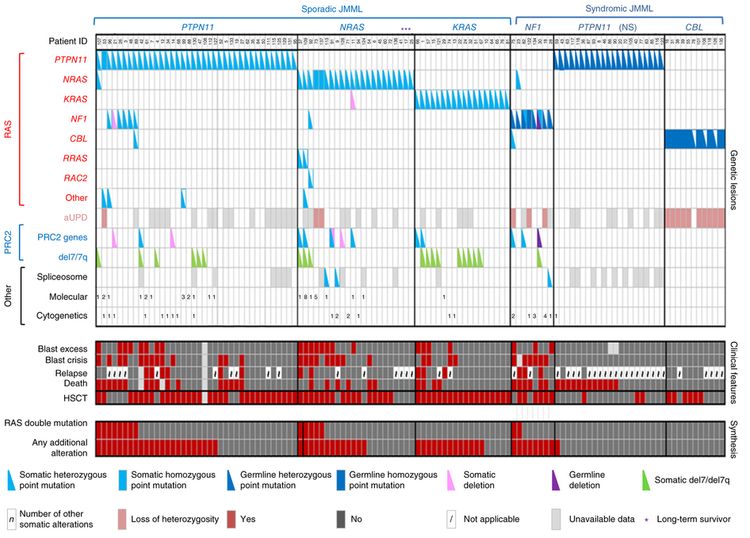 Figure 5. Alteration profiles in individual JMML cases. Germline and somatically acquired alterations with recurring hits in the RAS pathway and PRC2 network are shown for 118 patients with JMML who underwent detailed genetic analysis. Blast excess was defined as a blast count ≥10% but <20% of nucleated cells in the bone marrow at diagnosis. Blast crisis was defined as a blast count ≥20% of nucleated cells in the bone marrow. NS, Noonan syndrome. Reprinted by permission from Macmillan Publishers Ltd: Nature Genetics (Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47 [11]: 1334-40, 2015), copyright (2015).
Figure 5. Alteration profiles in individual JMML cases. Germline and somatically acquired alterations with recurring hits in the RAS pathway and PRC2 network are shown for 118 patients with JMML who underwent detailed genetic analysis. Blast excess was defined as a blast count ≥10% but <20% of nucleated cells in the bone marrow at diagnosis. Blast crisis was defined as a blast count ≥20% of nucleated cells in the bone marrow. NS, Noonan syndrome. Reprinted by permission from Macmillan Publishers Ltd: Nature Genetics (Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47 [11]: 1334-40, 2015), copyright (2015).
Genomic and Molecular Prognostic factors
Several genomic factors affect the prognosis of patients with JMML, including the following:
- Number of non–RAS pathway variants. A predictor of prognosis for children with JMML is the number of variants beyond the disease-defining RAS pathway variants.
- One study observed that zero or one somatic alteration (pathogenic variant or monosomy 7) was identified in 64 patients (65.3%) at diagnosis, whereas two or more alterations were identified in 34 patients (34.7%). In multivariate analysis, variant number (2 or more vs. 0 or 1) maintained significance as a predictor of inferior event-free survival (EFS) and overall survival (OS). A higher proportion of patients diagnosed with two or more alterations were older and male, and these patients also demonstrated a higher rate of monosomy 7 or somatic NF1 variants.
- Another study observed that approximately 60% of patients had one or more additional variants beyond their disease-defining RAS pathway variant. These patients had an inferior OS compared with patients who had no additional variants (3-year OS rate, 61% vs. 85%, respectively).
- A third study observed a trend for an inferior OS for patients with two or more variants compared with patients with zero or one variant.
- RAS pathway double variants. Although variants in the five canonical RAS pathway genes associated with JMML (NF1, NRAS, KRAS, PTPN11, and CBL) are generally mutually exclusive, 4% to 17% of cases have variants in two of these RAS pathway genes. This finding has been associated with a poorer prognosis.
- Two RAS pathway variants were identified in 11% of JMML patients in one report, and these patients had a significantly inferior EFS rate (14%) compared with patients who had a single RAS pathway variant (62%). Patients with Noonan syndrome were excluded from the analyses.
- Similar findings for RAS pathway variants were reported in a second study. This study observed that patients with RAS pathway double variants (15 of 96 patients) had lower survival rates than did patients with either no additional variants or with additional variants beyond the RAS pathway variant.
- DNA methylation profile.
- One study applied DNA methylation profiling to a discovery cohort of 39 patients with JMML and to a validation cohort of 40 patients. Distinctive subsets of JMML with either high, intermediate, or low methylation levels were observed in both cohorts. Patients with the lowest methylation levels had the highest survival rates, and all but 1 of 15 patients experienced spontaneous resolution in the low methylation cohort. High methylation status was associated with lower EFS rates.
- Another study applied DNA methylation profiling to a cohort of 106 patients with JMML. The study observed one subgroup of patients with a hypermethylation profile and one subgroup of patients with a hypomethylation profile. Patients in the hypermethylation group had a significantly lower OS rate than did patients in the hypomethylation group (5-year OS rate, 46% vs. 73%, respectively). Patients in the hypermethylation group also had a significantly poorer 5-year transplant-free survival rate than did patients in the hypomethylation group (2.2%; 95% CI, 0.2%–10.1% vs. 41.2%; 95% CI, 27.1%–54.8%). Hypermethylation status was associated with two or more variants, higher fetal hemoglobin levels, older age, and lower platelet count at diagnosis. All patients with Noonan syndrome were in the hypomethylation group.
- A study examined 33 patients with JMML who had CBL variants. The study identified 31 patients with low methylation and 2 patients with intermediate methylation. Both of the children with intermediate methylation relapsed after undergoing HSCT. Because treatment, which included observation only, varied among the 31 patients with low methylation, the impact of the methylation profile on therapeutic decisions and outcomes could not be fully assessed. However, the methylation status was not prognostic of spontaneous resolution.
- LIN28B overexpression.LIN28B overexpression, which is present in approximately one-half of children with JMML, identifies a biologically distinctive subset of JMML. LIN28B is an RNA-binding protein that regulates stem cell renewal.
- LIN28B overexpression was positively correlated with high blood fetal hemoglobin level and age (both of which are associated with poor prognosis), and it was negatively correlated with presence of monosomy 7 (also associated with inferior prognosis). Although LIN28B overexpression identifies a subset of patients with increased risk of treatment failure, it was not found to be an independent prognostic factor when other factors such as age and monosomy 7 status are considered.
- Another study also observed a subset of JMML patients with elevated LIN28B expression. The study identified LIN28B as the gene for which expression was most strongly associated with hypermethylation status.
For information about the treatment of JMML, see Juvenile Myelomonocytic Leukemia Treatment.
Myelodysplastic Neoplasms (MDS)
Molecular features of myelodysplastic neoplasms (MDS)
Compared with MDS in adults, pediatric MDS is associated with a distinctive constellation of genetic alterations. In adults, MDS often evolves from clonal hematopoiesis and is characterized by variants in TET2, DNMT3A, DDX41 and TP53. In contrast, variants in these genes are rare in pediatric MDS, while variants in GATA2, SAMD9, SAMD9L, ETV6, SETBP1, ASXL1, and RAS/MAPK pathway genes are observed in subsets of children with MDS.
A report of the genomic landscape of pediatric MDS described the results of whole-exome sequencing for 32 pediatric patients with primary MDS and targeted sequencing for another 14 cases. These 46 cases were equally divided between childhood MDS with low blasts (cMDS-LB) (previously called refractory cytopenia of childhood) and childhood MDS with increased blasts (cMDS-IB) (previously called MDS with excess blasts [MDS-EB)]). The results from the report include the following:
- Variants in RAS/MAPK pathway genes were observed in 43% of primary MDS cases, with variants most commonly involving the PTPN11 and NRAS genes. However, variants were also observed in other RAS/MAPK pathway genes (e.g., BRAF [non–BRAF V600E], CBL, and KRAS). RAS/MAPK variants were more common in patients with cMDS-IB (65%) than in patients with cMDS-LB (17%).
- Germline pathogenic variants in SAMD9 (n = 4) or SAMD9L (n = 4) were observed in 17% of patients with primary MDS, with seven of eight variants occurring in patients with cMDS-LB. These cases all showed loss of material on chromosome 7. Approximately 40% of patients with deletions of part or all of chromosome 7 had germline SAMD9 or SAMD9L variants.
- GATA2 pathogenic variants were observed in three cases (7%), and all cases were confirmed or presumed to be germline.
- Deletions involving chromosome 7 were the most common copy number alteration and were observed in 41% of cases. Loss of part or all of chromosome 7 was most commonly observed in SAMD9 and SAMD9L cases (100%) and in cMDS-IB patients with a RAS/MAPK variant (71%).
- In more than 1 of the 46 cases, other genes were altered (SETBP1, ETV6, and TP53).
A second report described the application of a targeted sequencing panel of 105 genes to 50 pediatric patients with MDS (cMDS-LB = 31 and cMDS-IB = 19) and was enriched for cases with monosomy 7 (48%). SAMD9 and SAMD9L were not included in the gene panel. The second report described the following results:
- Germline GATA2 pathogenic variants were observed in 30% of patients, and germline RUNX1 pathogenic variants were observed in 6% of patients.
- Somatic variants were observed in 34% of patients and were more common in patients with cMDS-IB than in patients with cMDS-LB (68% vs. 13%).
- The most commonly altered gene was SETBP1 (18%). Less commonly altered genes included ASXL1, RUNX1, and RAS/MAPK pathway genes (PTPN11, NRAS, KRAS, NF1). Twelve percent of cases had variants in RAS/MAPK pathway genes.
Patients with germline GATA2 pathogenic variants, in addition to MDS, show a wide range of hematopoietic and immune defects as well as nonhematopoietic manifestations. The former defects include monocytopenia with susceptibility to atypical mycobacterial infection and DCML deficiency (loss of dendritic cells, monocytes, and B and natural killer lymphoid cells). The resulting immunodeficiency leads to increased susceptibility to warts, severe viral infections, mycobacterial infections, fungal infections, and human papillomavirus–related cancers. The nonhematopoietic manifestations include deafness and lymphedema.
Germline GATA2 pathogenic variants were studied in 426 pediatric patients with primary MDS and 82 cases with secondary MDS who were enrolled in consecutive studies of the European Working Group of MDS in Childhood (EWOG-MDS). The study had the following results:
- Germline GATA2 pathogenic variants were identified in 7% of pediatric patients with primary MDS. While the median age of patients presenting with GATA2 variants was 12.3 years in the EWOG-MDS pediatric population, most cases of germline GATA2-related myeloid neoplasms occur during adulthood.
- GATA2 variants were more common in patients with cMDS-IB (15%) than in patients with cMDS-LB (4%).
- Among patients with GATA2 variants, 46% presented with cMDS-IB, and 70% showed monosomy 7.
- Familial MDS/acute myeloid leukemia (AML) was identified in 12 of 53 patients with GATA2 variants for whom detailed family histories were available.
- Nonhematologic phenotypes of GATA2 deficiency were present in 51% of patients with MDS who had GATA2 variants and included deafness (9%), lymphedema/hydrocele (23%), and immunodeficiency (39%).
SAMD9 and SAMD9L germline pathogenic variants are both associated with pediatric MDS cases in which there is an additional loss of all or part of chromosome 7.
In 2016, SAMD9 was identified as the cause of the MIRAGE syndrome (myelodysplasia, infection, restriction of growth, adrenal hypoplasia, genital phenotypes, and enteropathy), which is associated with early-onset MDS with monosomy 7. Subsequently, variants in SAMD9L were identified in patients with ataxia pancytopenia syndrome (ATXPC; OMIM 159550). SAMD9 and SAMD9L variants were also identified as the cause of the myelodysplasia and leukemia syndrome with monosomy 7 (MLSM7; OMIM 252270), a syndrome first identified in phenotypically normal siblings who developed MDS or AML associated with monosomy 7 during childhood.
- Causative variants in both SAMD9 and SAMD9L are gain-of-function variants and enhance the growth-suppressing activity of SAMD9 and SAMD9L.
- Both SAMD9 and SAMD9L are located at chromosome 7q21.2. Cases of MDS in patients with SAMD9 or SAMD9L variants often show monosomy 7, with the remaining chromosome 7 having wild-type SAMD9 and SAMD9L. This results in the loss of the enhanced growth-suppressing activity of the altered gene.
- Phenotypically normal patients with SAMD9 or SAMD9L variants and monosomy 7 may progress to develop MDS or AML or, alternatively, may show loss of their monosomy 7 with a return of normal hematopoiesis. The former outcome is associated with the acquisition of variants in genes associated with MDS/AML (e.g., ETV6 or SETBP1). The latter outcome is associated with genetic alterations (e.g., revertant variants or copy-neutral loss of heterozygosity with retention of the wild-type allele) that result in normalization of SAMD9 or SAMD9L activity. These observations suggest that monitoring patients with SAMD9- or SAMD9L-related monosomy 7, using clinical sequencing for acquired somatic variants in genes associated with progression to AML, may identify those at high risk of leukemic transformation. Such patients may benefit most from hematopoietic stem cell transplant.
The presence of an isolated monosomy 7 is the most common cytogenetic abnormality, although it does not appear to portend a poor prognosis, compared with its presence in overt AML. However, the presence of monosomy 7 in combination with other cytogenetic abnormalities is associated with a poor prognosis. The relatively common abnormalities of -Y, 20q-, and 5q- in adults with MDS are rare in childhood MDS. The presence of cytogenetic abnormalities that are found in AML (t(8;21)(q22;q22.1), inv(16)(p13.1;q22) or t(16;16)(p13.1;q22), and APL with PML::RARA gene fusions) defines disease that should be treated as AML and not MDS, regardless of blast percentage. The World Health Organization (WHO) notes that whether this should also apply to other recurring genetic abnormalities remains controversial.
For information about the treatment of childhood MDS, see Childhood Myelodysplastic Neoplasms Treatment.
Transient Abnormal Myelopoiesis (TAM)
Genomics of TAM
TAM blasts most commonly have megakaryoblastic differentiation characteristics and distinctive variants involving the GATA1 gene in the presence of trisomy 21. TAM may occur in phenotypically normal infants with genetic mosaicism in the bone marrow for trisomy 21. While TAM is generally not characterized by cytogenetic abnormalities other than trisomy 21, the presence of additional cytogenetic findings may predict an increased risk of developing subsequent AML.
GATA1 variants are present in most, if not all, children with Down syndrome who have either transient abnormal myelopoiesis (TAM) or acute megakaryoblastic leukemia (AMKL). GATA1 is a transcription factor that is required for normal development of erythroid cells, megakaryocytes, eosinophils, and mast cells. X-linked GATA1 variants result in the absence of the full-length GATA1 protein, leaving only the normally minor variant, a truncated GATA1s transcription factor that has decreased activity. This confers increased sensitivity to cytarabine by down-regulating cytidine deaminase expression, possibly explaining the superior outcome of children with Down syndrome and M7 AML when treated with cytarabine-containing regimens.
Approximately 20% of infants with TAM and Down syndrome eventually develop AML. Most of these cases are diagnosed within the first 3 years of life.
For information about the treatment of TAM, see Childhood Myeloid Proliferations Associated With Down Syndrome Treatment.
References
- Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet 54 (9): 1376-1389, 2022.
- Mullighan CG, Goorha S, Radtke I, et al.: Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446 (7137): 758-64, 2007.
- Mullighan CG, Miller CB, Radtke I, et al.: BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 453 (7191): 110-4, 2008.
- Roberts KG, Li Y, Payne-Turner D, et al.: Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 371 (11): 1005-15, 2014.
- Lilljebjörn H, Henningsson R, Hyrenius-Wittsten A, et al.: Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun 7: 11790, 2016.
- Zhang J, McCastlain K, Yoshihara H, et al.: Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet 48 (12): 1481-1489, 2016.
- Holmfeldt L, Wei L, Diaz-Flores E, et al.: The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 45 (3): 242-52, 2013.
- Loh ML, Zhang J, Harvey RC, et al.: Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children's Oncology Group TARGET Project. Blood 121 (3): 485-8, 2013.
- Bercovich D, Ganmore I, Scott LM, et al.: Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet 372 (9648): 1484-92, 2008.
- Li Z, Chang TC, Junco JJ, et al.: Genomic landscape of Down syndrome-associated acute lymphoblastic leukemia. Blood 142 (2): 172-184, 2023.
- Andersson AK, Ma J, Wang J, et al.: The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet 47 (4): 330-7, 2015.
- Ma X, Edmonson M, Yergeau D, et al.: Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun 6: 6604, 2015.
- Meyer JA, Wang J, Hogan LE, et al.: Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet 45 (3): 290-4, 2013.
- Li B, Li H, Bai Y, et al.: Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat Med 21 (6): 563-71, 2015.
- Mullighan CG, Zhang J, Kasper LH, et al.: CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471 (7337): 235-9, 2011.
- Mattano LA, Devidas M, Maloney KW, et al.: Favorable Trisomies and ETV6-RUNX1 Predict Cure in Low-Risk B-Cell Acute Lymphoblastic Leukemia: Results From Children's Oncology Group Trial AALL0331. J Clin Oncol 39 (14): 1540-1552, 2021.
- Moorman AV, Ensor HM, Richards SM, et al.: Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol 11 (5): 429-38, 2010.
- Alaggio R, Amador C, Anagnostopoulos I, et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 36 (7): 1720-1748, 2022.
- Paulsson K, Johansson B: High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer 48 (8): 637-60, 2009.
- Aricò M, Valsecchi MG, Rizzari C, et al.: Long-term results of the AIEOP-ALL-95 Trial for Childhood Acute Lymphoblastic Leukemia: insight on the prognostic value of DNA index in the framework of Berlin-Frankfurt-Muenster based chemotherapy. J Clin Oncol 26 (2): 283-9, 2008.
- Dastugue N, Suciu S, Plat G, et al.: Hyperdiploidy with 58-66 chromosomes in childhood B-acute lymphoblastic leukemia is highly curable: 58951 CLG-EORTC results. Blood 121 (13): 2415-23, 2013.
- Synold TW, Relling MV, Boyett JM, et al.: Blast cell methotrexate-polyglutamate accumulation in vivo differs by lineage, ploidy, and methotrexate dose in acute lymphoblastic leukemia. J Clin Invest 94 (5): 1996-2001, 1994.
- Moorman AV, Richards SM, Martineau M, et al.: Outcome heterogeneity in childhood high-hyperdiploid acute lymphoblastic leukemia. Blood 102 (8): 2756-62, 2003.
- Chilton L, Buck G, Harrison CJ, et al.: High hyperdiploidy among adolescents and adults with acute lymphoblastic leukaemia (ALL): cytogenetic features, clinical characteristics and outcome. Leukemia 28 (7): 1511-8, 2014.
- Sutcliffe MJ, Shuster JJ, Sather HN, et al.: High concordance from independent studies by the Children's Cancer Group (CCG) and Pediatric Oncology Group (POG) associating favorable prognosis with combined trisomies 4, 10, and 17 in children with NCI Standard-Risk B-precursor Acute Lymphoblastic Leukemia: a Children's Oncology Group (COG) initiative. Leukemia 19 (5): 734-40, 2005.
- Harris MB, Shuster JJ, Carroll A, et al.: Trisomy of leukemic cell chromosomes 4 and 10 identifies children with B-progenitor cell acute lymphoblastic leukemia with a very low risk of treatment failure: a Pediatric Oncology Group study. Blood 79 (12): 3316-24, 1992.
- Enshaei A, Vora A, Harrison CJ, et al.: Defining low-risk high hyperdiploidy in patients with paediatric acute lymphoblastic leukaemia: a retrospective analysis of data from the UKALL97/99 and UKALL2003 clinical trials. Lancet Haematol 8 (11): e828-e839, 2021.
- Heerema NA, Harbott J, Galimberti S, et al.: Secondary cytogenetic aberrations in childhood Philadelphia chromosome positive acute lymphoblastic leukemia are nonrandom and may be associated with outcome. Leukemia 18 (4): 693-702, 2004.
- Carroll AJ, Shago M, Mikhail FM, et al.: Masked hypodiploidy: Hypodiploid acute lymphoblastic leukemia (ALL) mimicking hyperdiploid ALL in children: A report from the Children's Oncology Group. Cancer Genet 238: 62-68, 2019.
- Nachman JB, Heerema NA, Sather H, et al.: Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood 110 (4): 1112-5, 2007.
- Raimondi SC, Zhou Y, Shurtleff SA, et al.: Near-triploidy and near-tetraploidy in childhood acute lymphoblastic leukemia: association with B-lineage blast cells carrying the ETV6-RUNX1 fusion, T-lineage immunophenotype, and favorable outcome. Cancer Genet Cytogenet 169 (1): 50-7, 2006.
- Attarbaschi A, Mann G, König M, et al.: Incidence and relevance of secondary chromosome abnormalities in childhood TEL/AML1+ acute lymphoblastic leukemia: an interphase FISH analysis. Leukemia 18 (10): 1611-6, 2004.
- Lemez P, Attarbaschi A, Béné MC, et al.: Childhood near-tetraploid acute lymphoblastic leukemia: an EGIL study on 36 cases. Eur J Haematol 85 (4): 300-8, 2010.
- Paulsson K, Lilljebjörn H, Biloglav A, et al.: The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet 47 (6): 672-6, 2015.
- Harrison CJ, Moorman AV, Broadfield ZJ, et al.: Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol 125 (5): 552-9, 2004.
- Mullighan CG, Jeha S, Pei D, et al.: Outcome of children with hypodiploid ALL treated with risk-directed therapy based on MRD levels. Blood 126 (26): 2896-9, 2015.
- Pui CH, Rebora P, Schrappe M, et al.: Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Retrospective Multinational Study. J Clin Oncol 37 (10): 770-779, 2019.
- McNeer JL, Devidas M, Dai Y, et al.: Hematopoietic Stem-Cell Transplantation Does Not Improve the Poor Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Report From Children's Oncology Group. J Clin Oncol 37 (10): 780-789, 2019.
- Irving J, Matheson E, Minto L, et al.: Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood 124 (23): 3420-30, 2014.
- Qian M, Cao X, Devidas M, et al.: TP53 Germline Variations Influence the Predisposition and Prognosis of B-Cell Acute Lymphoblastic Leukemia in Children. J Clin Oncol 36 (6): 591-599, 2018.
- Rubnitz JE, Wichlan D, Devidas M, et al.: Prospective analysis of TEL gene rearrangements in childhood acute lymphoblastic leukemia: a Children's Oncology Group study. J Clin Oncol 26 (13): 2186-91, 2008.
- Kanerva J, Saarinen-Pihkala UM, Niini T, et al.: Favorable outcome in 20-year follow-up of children with very-low-risk ALL and minimal standard therapy, with special reference to TEL-AML1 fusion. Pediatr Blood Cancer 42 (1): 30-5, 2004.
- Aldrich MC, Zhang L, Wiemels JL, et al.: Cytogenetics of Hispanic and White children with acute lymphoblastic leukemia in California. Cancer Epidemiol Biomarkers Prev 15 (3): 578-81, 2006.
- Loh ML, Goldwasser MA, Silverman LB, et al.: Prospective analysis of TEL/AML1-positive patients treated on Dana-Farber Cancer Institute Consortium Protocol 95-01. Blood 107 (11): 4508-13, 2006.
- Borowitz MJ, Devidas M, Hunger SP, et al.: Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood 111 (12): 5477-85, 2008.
- Madzo J, Zuna J, Muzíková K, et al.: Slower molecular response to treatment predicts poor outcome in patients with TEL/AML1 positive acute lymphoblastic leukemia: prospective real-time quantitative reverse transcriptase-polymerase chain reaction study. Cancer 97 (1): 105-13, 2003.
- Bhojwani D, Pei D, Sandlund JT, et al.: ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: improved outcome with contemporary therapy. Leukemia 26 (2): 265-70, 2012.
- Enshaei A, Schwab CJ, Konn ZJ, et al.: Long-term follow-up of ETV6-RUNX1 ALL reveals that NCI risk, rather than secondary genetic abnormalities, is the key risk factor. Leukemia 27 (11): 2256-9, 2013.
- Barbany G, Andersen MK, Autio K, et al.: Additional aberrations of the ETV6 and RUNX1 genes have no prognostic impact in 229 t(12;21)(p13;q22)-positive B-cell precursor acute lymphoblastic leukaemias treated according to the NOPHO-ALL-2000 protocol. Leuk Res 36 (7): 936-8, 2012.
- Forestier E, Heyman M, Andersen MK, et al.: Outcome of ETV6/RUNX1-positive childhood acute lymphoblastic leukaemia in the NOPHO-ALL-1992 protocol: frequent late relapses but good overall survival. Br J Haematol 140 (6): 665-72, 2008.
- Seeger K, Stackelberg AV, Taube T, et al.: Relapse of TEL-AML1--positive acute lymphoblastic leukemia in childhood: a matched-pair analysis. J Clin Oncol 19 (13): 3188-93, 2001.
- Gandemer V, Chevret S, Petit A, et al.: Excellent prognosis of late relapses of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia: lessons from the FRALLE 93 protocol. Haematologica 97 (11): 1743-50, 2012.
- Zuna J, Ford AM, Peham M, et al.: TEL deletion analysis supports a novel view of relapse in childhood acute lymphoblastic leukemia. Clin Cancer Res 10 (16): 5355-60, 2004.
- van Delft FW, Horsley S, Colman S, et al.: Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 117 (23): 6247-54, 2011.
- Aricò M, Schrappe M, Hunger SP, et al.: Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol 28 (31): 4755-61, 2010.
- Schrappe M, Aricò M, Harbott J, et al.: Philadelphia chromosome-positive (Ph+) childhood acute lymphoblastic leukemia: good initial steroid response allows early prediction of a favorable treatment outcome. Blood 92 (8): 2730-41, 1998.
- Ribeiro RC, Broniscer A, Rivera GK, et al.: Philadelphia chromosome-positive acute lymphoblastic leukemia in children: durable responses to chemotherapy associated with low initial white blood cell counts. Leukemia 11 (9): 1493-6, 1997.
- Biondi A, Schrappe M, De Lorenzo P, et al.: Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol 13 (9): 936-45, 2012.
- Schultz KR, Bowman WP, Aledo A, et al.: Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol 27 (31): 5175-81, 2009.
- Schultz KR, Carroll A, Heerema NA, et al.: Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children's Oncology Group study AALL0031. Leukemia 28 (7): 1467-71, 2014.
- Duffield AS, Mullighan CG, Borowitz MJ: International Consensus Classification of acute lymphoblastic leukemia/lymphoma. Virchows Arch 482 (1): 11-26, 2023.
- Short NJ, Jabbour E, Macaron W, et al.: Ultrasensitive NGS MRD assessment in Ph+ ALL: Prognostic impact and correlation with RT-PCR for BCR::ABL1. Am J Hematol 98 (8): 1196-1203, 2023.
- Hovorkova L, Zaliova M, Venn NC, et al.: Monitoring of childhood ALL using BCR-ABL1 genomic breakpoints identifies a subgroup with CML-like biology. Blood 129 (20): 2771-2781, 2017.
- Zuna J, Hovorkova L, Krotka J, et al.: Minimal residual disease in BCR::ABL1-positive acute lymphoblastic leukemia: different significance in typical ALL and in CML-like disease. Leukemia 36 (12): 2793-2801, 2022.
- Hunger SP, Tran TH, Saha V, et al.: Dasatinib with intensive chemotherapy in de novo paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (CA180-372/COG AALL1122): a single-arm, multicentre, phase 2 trial. Lancet Haematol 10 (7): e510-e520, 2023.
- Pui CH, Chessells JM, Camitta B, et al.: Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia 17 (4): 700-6, 2003.
- Johansson B, Moorman AV, Haas OA, et al.: Hematologic malignancies with t(4;11)(q21;q23)--a cytogenetic, morphologic, immunophenotypic and clinical study of 183 cases. European 11q23 Workshop participants. Leukemia 12 (5): 779-87, 1998.
- Raimondi SC, Peiper SC, Kitchingman GR, et al.: Childhood acute lymphoblastic leukemia with chromosomal breakpoints at 11q23. Blood 73 (6): 1627-34, 1989.
- Harrison CJ, Moorman AV, Barber KE, et al.: Interphase molecular cytogenetic screening for chromosomal abnormalities of prognostic significance in childhood acute lymphoblastic leukaemia: a UK Cancer Cytogenetics Group Study. Br J Haematol 129 (4): 520-30, 2005.
- Pui CH, Pei D, Campana D, et al.: A revised definition for cure of childhood acute lymphoblastic leukemia. Leukemia 28 (12): 2336-43, 2014.
- Pieters R, Schrappe M, De Lorenzo P, et al.: A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 370 (9583): 240-50, 2007.
- Attarbaschi A, Möricke A, Harrison CJ, et al.: Outcomes of Childhood Noninfant Acute Lymphoblastic Leukemia With 11q23/KMT2A Rearrangements in a Modern Therapy Era: A Retrospective International Study. J Clin Oncol 41 (7): 1404-1422, 2023.
- Isobe T, Takagi M, Sato-Otsubo A, et al.: Multi-omics analysis defines highly refractory RAS burdened immature subgroup of infant acute lymphoblastic leukemia. Nat Commun 13 (1): 4501, 2022.
- Pui CH, Gaynon PS, Boyett JM, et al.: Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet 359 (9321): 1909-15, 2002.
- Rubnitz JE, Camitta BM, Mahmoud H, et al.: Childhood acute lymphoblastic leukemia with the MLL-ENL fusion and t(11;19)(q23;p13.3) translocation. J Clin Oncol 17 (1): 191-6, 1999.
- Hunger SP: Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood 87 (4): 1211-24, 1996.
- Uckun FM, Sensel MG, Sather HN, et al.: Clinical significance of translocation t(1;19) in childhood acute lymphoblastic leukemia in the context of contemporary therapies: a report from the Children's Cancer Group. J Clin Oncol 16 (2): 527-35, 1998.
- Fischer U, Forster M, Rinaldi A, et al.: Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet 47 (9): 1020-9, 2015.
- Pui CH, Sandlund JT, Pei D, et al.: Results of therapy for acute lymphoblastic leukemia in black and white children. JAMA 290 (15): 2001-7, 2003.
- Crist WM, Carroll AJ, Shuster JJ, et al.: Poor prognosis of children with pre-B acute lymphoblastic leukemia is associated with the t(1;19)(q23;p13): a Pediatric Oncology Group study. Blood 76 (1): 117-22, 1990.
- Andersen MK, Autio K, Barbany G, et al.: Paediatric B-cell precursor acute lymphoblastic leukaemia with t(1;19)(q23;p13): clinical and cytogenetic characteristics of 47 cases from the Nordic countries treated according to NOPHO protocols. Br J Haematol 155 (2): 235-43, 2011.
- Pui CH, Campana D, Pei D, et al.: Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med 360 (26): 2730-41, 2009.
- Jeha S, Pei D, Raimondi SC, et al.: Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia 23 (8): 1406-9, 2009.
- Minson KA, Prasad P, Vear S, et al.: t(17;19) in Children with Acute Lymphocytic Leukemia: A Report of 3 Cases and a Review of the Literature. Case Rep Hematol 2013: 563291, 2013.
- Lee SHR, Antillon-Klussmann F, Pei D, et al.: Association of Genetic Ancestry With the Molecular Subtypes and Prognosis of Childhood Acute Lymphoblastic Leukemia. JAMA Oncol 8 (3): 354-363, 2022.
- Zaliova M, Potuckova E, Hovorkova L, et al.: ERG deletions in childhood acute lymphoblastic leukemia with DUX4 rearrangements are mostly polyclonal, prognostically relevant and their detection rate strongly depends on screening method sensitivity. Haematologica 104 (7): 1407-1416, 2019.
- Harvey RC, Mullighan CG, Wang X, et al.: Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood 116 (23): 4874-84, 2010.
- Zaliova M, Zimmermannova O, Dörge P, et al.: ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia 28 (1): 182-5, 2014.
- Clappier E, Auclerc MF, Rapion J, et al.: An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia 28 (1): 70-7, 2014.
- Li Z, Lee SHR, Chin WHN, et al.: Distinct clinical characteristics of DUX4- and PAX5-altered childhood B-lymphoblastic leukemia. Blood Adv 5 (23): 5226-5238, 2021.
- Gu Z, Churchman M, Roberts K, et al.: Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun 7: 13331, 2016.
- Liu YF, Wang BY, Zhang WN, et al.: Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 8: 173-83, 2016.
- Suzuki K, Okuno Y, Kawashima N, et al.: MEF2D-BCL9 Fusion Gene Is Associated With High-Risk Acute B-Cell Precursor Lymphoblastic Leukemia in Adolescents. J Clin Oncol 34 (28): 3451-9, 2016.
- Lilljebjörn H, Ågerstam H, Orsmark-Pietras C, et al.: RNA-seq identifies clinically relevant fusion genes in leukemia including a novel MEF2D/CSF1R fusion responsive to imatinib. Leukemia 28 (4): 977-9, 2014.
- Hirabayashi S, Ohki K, Nakabayashi K, et al.: ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 102 (1): 118-129, 2017.
- Qian M, Zhang H, Kham SK, et al.: Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res 27 (2): 185-195, 2017.
- Hirabayashi S, Butler ER, Ohki K, et al.: Clinical characteristics and outcomes of B-ALL with ZNF384 rearrangements: a retrospective analysis by the Ponte di Legno Childhood ALL Working Group. Leukemia 35 (11): 3272-3277, 2021.
- Shago M, Abla O, Hitzler J, et al.: Frequency and outcome of pediatric acute lymphoblastic leukemia with ZNF384 gene rearrangements including a novel translocation resulting in an ARID1B/ZNF384 gene fusion. Pediatr Blood Cancer 63 (11): 1915-21, 2016.
- Yao L, Cen J, Pan J, et al.: TAF15-ZNF384 fusion gene in childhood mixed phenotype acute leukemia. Cancer Genet 211: 1-4, 2017.
- Alexander TB, Gu Z, Iacobucci I, et al.: The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 562 (7727): 373-379, 2018.
- Boer JM, Valsecchi MG, Hormann FM, et al.: Favorable outcome of NUTM1-rearranged infant and pediatric B cell precursor acute lymphoblastic leukemia in a collaborative international study. Leukemia 35 (10): 2978-2982, 2021.
- De Lorenzo P, Moorman AV, Pieters R, et al.: Cytogenetics and outcome of infants with acute lymphoblastic leukemia and absence of MLL rearrangements. Leukemia 28 (2): 428-30, 2014.
- Fazio G, Bardini M, De Lorenzo P, et al.: Recurrent genetic fusions redefine MLL germ line acute lymphoblastic leukemia in infants. Blood 137 (14): 1980-1984, 2021.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Hogan TF, Koss W, Murgo AJ, et al.: Acute lymphoblastic leukemia with chromosomal 5;14 translocation and hypereosinophilia: case report and literature review. J Clin Oncol 5 (3): 382-90, 1987.
- Grimaldi JC, Meeker TC: The t(5;14) chromosomal translocation in a case of acute lymphocytic leukemia joins the interleukin-3 gene to the immunoglobulin heavy chain gene. Blood 73 (8): 2081-5, 1989.
- Meeker TC, Hardy D, Willman C, et al.: Activation of the interleukin-3 gene by chromosome translocation in acute lymphocytic leukemia with eosinophilia. Blood 76 (2): 285-9, 1990.
- Sutton R, Lonergan M, Tapp H, et al.: Two cases of hypereosinophilia and high-risk acute lymphoblastic leukemia. Leukemia 22 (7): 1463-5, 2008.
- Koleilat A, Smadbeck JB, Zepeda-Mendoza CJ, et al.: Characterization of unusual iAMP21 B-lymphoblastic leukemia (iAMP21-ALL) from the Mayo Clinic and Children's Oncology Group. Genes Chromosomes Cancer 61 (12): 710-719, 2022.
- Heerema NA, Carroll AJ, Devidas M, et al.: Intrachromosomal amplification of chromosome 21 is associated with inferior outcomes in children with acute lymphoblastic leukemia treated in contemporary standard-risk children's oncology group studies: a report from the children's oncology group. J Clin Oncol 31 (27): 3397-402, 2013.
- Moorman AV, Robinson H, Schwab C, et al.: Risk-directed treatment intensification significantly reduces the risk of relapse among children and adolescents with acute lymphoblastic leukemia and intrachromosomal amplification of chromosome 21: a comparison of the MRC ALL97/99 and UKALL2003 trials. J Clin Oncol 31 (27): 3389-96, 2013.
- Harrison CJ, Moorman AV, Schwab C, et al.: An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia 28 (5): 1015-21, 2014.
- Gu Z, Churchman ML, Roberts KG, et al.: PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet 51 (2): 296-307, 2019.
- Strehl S, König M, Dworzak MN, et al.: PAX5/ETV6 fusion defines cytogenetic entity dic(9;12)(p13;p13). Leukemia 17 (6): 1121-3, 2003.
- Schwab C, Nebral K, Chilton L, et al.: Intragenic amplification of PAX5: a novel subgroup in B-cell precursor acute lymphoblastic leukemia? Blood Adv 1 (19): 1473-7, 2017.
- Den Boer ML, van Slegtenhorst M, De Menezes RX, et al.: A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 10 (2): 125-34, 2009.
- Mullighan CG, Su X, Zhang J, et al.: Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 360 (5): 470-80, 2009.
- Reshmi SC, Harvey RC, Roberts KG, et al.: Targetable kinase gene fusions in high-risk B-ALL: a study from the Children's Oncology Group. Blood 129 (25): 3352-3361, 2017.
- Roberts KG, Morin RD, Zhang J, et al.: Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 22 (2): 153-66, 2012.
- van der Veer A, Waanders E, Pieters R, et al.: Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood 122 (15): 2622-9, 2013.
- Roberts KG, Reshmi SC, Harvey RC, et al.: Genomic and outcome analyses of Ph-like ALL in NCI standard-risk patients: a report from the Children's Oncology Group. Blood 132 (8): 815-824, 2018.
- Roberts KG, Pei D, Campana D, et al.: Outcomes of children with BCR-ABL1–like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J Clin Oncol 32 (27): 3012-20, 2014.
- Harvey RC, Mullighan CG, Chen IM, et al.: Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 115 (26): 5312-21, 2010.
- Mullighan CG, Collins-Underwood JR, Phillips LA, et al.: Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41 (11): 1243-6, 2009.
- Schwab C, Roberts K, Boer JM, et al.: SSBP2-CSF1R is a recurrent fusion in B-lineage acute lymphoblastic leukemia with diverse genetic presentation and variable outcome. Blood 137 (13): 1835-1838, 2021.
- den Boer ML, Cario G, Moorman AV, et al.: Outcomes of paediatric patients with B-cell acute lymphocytic leukaemia with ABL-class fusion in the pre-tyrosine-kinase inhibitor era: a multicentre, retrospective, cohort study. Lancet Haematol 8 (1): e55-e66, 2021.
- Iacobucci I, Li Y, Roberts KG, et al.: Truncating Erythropoietin Receptor Rearrangements in Acute Lymphoblastic Leukemia. Cancer Cell 29 (2): 186-200, 2016.
- Cario G, Zimmermann M, Romey R, et al.: Presence of the P2RY8-CRLF2 rearrangement is associated with a poor prognosis in non-high-risk precursor B-cell acute lymphoblastic leukemia in children treated according to the ALL-BFM 2000 protocol. Blood 115 (26): 5393-7, 2010.
- Ensor HM, Schwab C, Russell LJ, et al.: Demographic, clinical, and outcome features of children with acute lymphoblastic leukemia and CRLF2 deregulation: results from the MRC ALL97 clinical trial. Blood 117 (7): 2129-36, 2011.
- Schmäh J, Fedders B, Panzer-Grümayer R, et al.: Molecular characterization of acute lymphoblastic leukemia with high CRLF2 gene expression in childhood. Pediatr Blood Cancer 64 (10): , 2017.
- Raca G, Abdel-Azim H, Yue F, et al.: Increased Incidence of IKZF1 deletions and IGH-CRLF2 translocations in B-ALL of Hispanic/Latino children-a novel health disparity. Leukemia 35 (8): 2399-2402, 2021.
- Vesely C, Frech C, Eckert C, et al.: Genomic and transcriptional landscape of P2RY8-CRLF2-positive childhood acute lymphoblastic leukemia. Leukemia 31 (7): 1491-1501, 2017.
- Russell LJ, Jones L, Enshaei A, et al.: Characterisation of the genomic landscape of CRLF2-rearranged acute lymphoblastic leukemia. Genes Chromosomes Cancer 56 (5): 363-372, 2017.
- Potter N, Jones L, Blair H, et al.: Single-cell analysis identifies CRLF2 rearrangements as both early and late events in Down syndrome and non-Down syndrome acute lymphoblastic leukaemia. Leukemia 33 (4): 893-904, 2019.
- Morak M, Attarbaschi A, Fischer S, et al.: Small sizes and indolent evolutionary dynamics challenge the potential role of P2RY8-CRLF2-harboring clones as main relapse-driving force in childhood ALL. Blood 120 (26): 5134-42, 2012.
- Schwab CJ, Chilton L, Morrison H, et al.: Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica 98 (7): 1081-8, 2013.
- Chen IM, Harvey RC, Mullighan CG, et al.: Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 119 (15): 3512-22, 2012.
- Palmi C, Vendramini E, Silvestri D, et al.: Poor prognosis for P2RY8-CRLF2 fusion but not for CRLF2 over-expression in children with intermediate risk B-cell precursor acute lymphoblastic leukemia. Leukemia 26 (10): 2245-53, 2012.
- Clappier E, Grardel N, Bakkus M, et al.: IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: results of the EORTC Children's Leukemia Group study 58951. Leukemia 29 (11): 2154-61, 2015.
- Srinivasan S, Ramanathan S, Kumar S, et al.: Prevalence and prognostic significance of IKZF1 deletion in paediatric acute lymphoblastic leukemia: A systematic review and meta-analysis. Ann Hematol 102 (8): 2165-2179, 2023.
- Buitenkamp TD, Pieters R, Gallimore NE, et al.: Outcome in children with Down's syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia 26 (10): 2204-11, 2012.
- Krentz S, Hof J, Mendioroz A, et al.: Prognostic value of genetic alterations in children with first bone marrow relapse of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia 27 (2): 295-304, 2013.
- Feng J, Tang Y: Prognostic significance of IKZF1 alteration status in pediatric B-lineage acute lymphoblastic leukemia: a meta-analysis. Leuk Lymphoma 54 (4): 889-91, 2013.
- Dörge P, Meissner B, Zimmermann M, et al.: IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica 98 (3): 428-32, 2013.
- Olsson L, Castor A, Behrendtz M, et al.: Deletions of IKZF1 and SPRED1 are associated with poor prognosis in a population-based series of pediatric B-cell precursor acute lymphoblastic leukemia diagnosed between 1992 and 2011. Leukemia 28 (2): 302-10, 2014.
- Boer JM, van der Veer A, Rizopoulos D, et al.: Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia 30 (1): 32-8, 2016.
- Tran TH, Harris MH, Nguyen JV, et al.: Prognostic impact of kinase-activating fusions and IKZF1 deletions in pediatric high-risk B-lineage acute lymphoblastic leukemia. Blood Adv 2 (5): 529-533, 2018.
- Vrooman LM, Blonquist TM, Harris MH, et al.: Refining risk classification in childhood B acute lymphoblastic leukemia: results of DFCI ALL Consortium Protocol 05-001. Blood Adv 2 (12): 1449-1458, 2018.
- Öfverholm I, Rezayee F, Heyman M, et al.: The prognostic impact of IKZF1 deletions and UKALL genetic classifiers in paediatric B-cell precursor acute lymphoblastic leukaemia treated according to NOPHO 2008 protocols. Br J Haematol 202 (2): 384-392, 2023.
- van der Veer A, Zaliova M, Mottadelli F, et al.: IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood 123 (11): 1691-8, 2014.
- Stanulla M, Dagdan E, Zaliova M, et al.: IKZF1plus Defines a New Minimal Residual Disease-Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J Clin Oncol 36 (12): 1240-1249, 2018.
- Mangum DS, Meyer JA, Mason CC, et al.: Association of Combined Focal 22q11.22 Deletion and IKZF1 Alterations With Outcomes in Childhood Acute Lymphoblastic Leukemia. JAMA Oncol 7 (10): 1521-1528, 2021.
- Yeoh AEJ, Lu Y, Chin WHN, et al.: Intensifying Treatment of Childhood B-Lymphoblastic Leukemia With IKZF1 Deletion Reduces Relapse and Improves Overall Survival: Results of Malaysia-Singapore ALL 2010 Study. J Clin Oncol 36 (26): 2726-2735, 2018.
- Pieters R, de Groot-Kruseman H, Fiocco M, et al.: Improved Outcome for ALL by Prolonging Therapy for IKZF1 Deletion and Decreasing Therapy for Other Risk Groups. J Clin Oncol 41 (25): 4130-4142, 2023.
- Herbrueggen H, Mueller S, Rohde J, et al.: Treatment and outcome of IG-MYC+ neoplasms with precursor B-cell phenotype in childhood and adolescence. Leukemia 34 (3): 942-946, 2020.
- Sakaguchi K, Imamura T, Ishimaru S, et al.: Nationwide study of pediatric B-cell precursor acute lymphoblastic leukemia with chromosome 8q24/MYC rearrangement in Japan. Pediatr Blood Cancer 67 (7): e28341, 2020.
- Bomken S, Enshaei A, Schwalbe EC, et al.: Molecular characterization and clinical outcome of B-cell precursor acute lymphoblastic leukemia with IG-MYC rearrangement. Haematologica 108 (3): 717-731, 2023.
- Liu Y, Easton J, Shao Y, et al.: The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet 49 (8): 1211-1218, 2017.
- Armstrong SA, Look AT: Molecular genetics of acute lymphoblastic leukemia. J Clin Oncol 23 (26): 6306-15, 2005.
- Karrman K, Forestier E, Heyman M, et al.: Clinical and cytogenetic features of a population-based consecutive series of 285 pediatric T-cell acute lymphoblastic leukemias: rare T-cell receptor gene rearrangements are associated with poor outcome. Genes Chromosomes Cancer 48 (9): 795-805, 2009.
- Mansour MR, Abraham BJ, Anders L, et al.: Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346 (6215): 1373-7, 2014.
- Steimlé T, Dourthe ME, Alcantara M, et al.: Clinico-biological features of T-cell acute lymphoblastic leukemia with fusion proteins. Blood Cancer J 12 (1): 14, 2022.
- Weng AP, Ferrando AA, Lee W, et al.: Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306 (5694): 269-71, 2004.
- Gallo Llorente L, Luther H, Schneppenheim R, et al.: Identification of novel NOTCH1 mutations: increasing our knowledge of the NOTCH signaling pathway. Pediatr Blood Cancer 61 (5): 788-96, 2014.
- Burns MA, Place AE, Stevenson KE, et al.: Identification of prognostic factors in childhood T-cell acute lymphoblastic leukemia: Results from DFCI ALL Consortium Protocols 05-001 and 11-001. Pediatr Blood Cancer 68 (1): e28719, 2021.
- Petit A, Trinquand A, Chevret S, et al.: Oncogenetic mutations combined with MRD improve outcome prediction in pediatric T-cell acute lymphoblastic leukemia. Blood 131 (3): 289-300, 2018.
- Trinquand A, Tanguy-Schmidt A, Ben Abdelali R, et al.: Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: a Group for Research in Adult Acute Lymphoblastic Leukemia study. J Clin Oncol 31 (34): 4333-42, 2013.
- Bergeron J, Clappier E, Radford I, et al.: Prognostic and oncogenic relevance of TLX1/HOX11 expression level in T-ALLs. Blood 110 (7): 2324-30, 2007.
- van Grotel M, Meijerink JP, Beverloo HB, et al.: The outcome of molecular-cytogenetic subgroups in pediatric T-cell acute lymphoblastic leukemia: a retrospective study of patients treated according to DCOG or COALL protocols. Haematologica 91 (9): 1212-21, 2006.
- Cavé H, Suciu S, Preudhomme C, et al.: Clinical significance of HOX11L2 expression linked to t(5;14)(q35;q32), of HOX11 expression, and of SIL-TAL fusion in childhood T-cell malignancies: results of EORTC studies 58881 and 58951. Blood 103 (2): 442-50, 2004.
- Baak U, Gökbuget N, Orawa H, et al.: Thymic adult T-cell acute lymphoblastic leukemia stratified in standard- and high-risk group by aberrant HOX11L2 expression: experience of the German multicenter ALL study group. Leukemia 22 (6): 1154-60, 2008.
- Ferrando AA, Neuberg DS, Dodge RK, et al.: Prognostic importance of TLX1 (HOX11) oncogene expression in adults with T-cell acute lymphoblastic leukaemia. Lancet 363 (9408): 535-6, 2004.
- Burmeister T, Gökbuget N, Reinhardt R, et al.: NUP214-ABL1 in adult T-ALL: the GMALL study group experience. Blood 108 (10): 3556-9, 2006.
- Graux C, Stevens-Kroef M, Lafage M, et al.: Heterogeneous patterns of amplification of the NUP214-ABL1 fusion gene in T-cell acute lymphoblastic leukemia. Leukemia 23 (1): 125-33, 2009.
- Hagemeijer A, Graux C: ABL1 rearrangements in T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer 49 (4): 299-308, 2010.
- Quintás-Cardama A, Tong W, Manshouri T, et al.: Activity of tyrosine kinase inhibitors against human NUP214-ABL1-positive T cell malignancies. Leukemia 22 (6): 1117-24, 2008.
- Clarke S, O'Reilly J, Romeo G, et al.: NUP214-ABL1 positive T-cell acute lymphoblastic leukemia patient shows an initial favorable response to imatinib therapy post relapse. Leuk Res 35 (7): e131-3, 2011.
- Deenik W, Beverloo HB, van der Poel-van de Luytgaarde SC, et al.: Rapid complete cytogenetic remission after upfront dasatinib monotherapy in a patient with a NUP214-ABL1-positive T-cell acute lymphoblastic leukemia. Leukemia 23 (3): 627-9, 2009.
- Crombet O, Lastrapes K, Zieske A, et al.: Complete morphologic and molecular remission after introduction of dasatinib in the treatment of a pediatric patient with t-cell acute lymphoblastic leukemia and ABL1 amplification. Pediatr Blood Cancer 59 (2): 333-4, 2012.
- Seki M, Kimura S, Isobe T, et al.: Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat Genet 49 (8): 1274-1281, 2017.
- Nagel S, Scherr M, Kel A, et al.: Activation of TLX3 and NKX2-5 in t(5;14)(q35;q32) T-cell acute lymphoblastic leukemia by remote 3'-BCL11B enhancers and coregulation by PU.1 and HMGA1. Cancer Res 67 (4): 1461-71, 2007.
- Gutierrez A, Kentsis A, Sanda T, et al.: The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood 118 (15): 4169-73, 2011.
- Ceppi F, Gotti G, Möricke A, et al.: Near-tetraploid T-cell acute lymphoblastic leukaemia in childhood: Results of the AIEOP-BFM ALL studies. Eur J Cancer 175: 120-124, 2022.
- Zhang J, Ding L, Holmfeldt L, et al.: The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481 (7380): 157-63, 2012.
- Gutierrez A, Dahlberg SE, Neuberg DS, et al.: Absence of biallelic TCRgamma deletion predicts early treatment failure in pediatric T-cell acute lymphoblastic leukemia. J Clin Oncol 28 (24): 3816-23, 2010.
- Yang YL, Hsiao CC, Chen HY, et al.: Absence of biallelic TCRγ deletion predicts induction failure and poorer outcomes in childhood T-cell acute lymphoblastic leukemia. Pediatr Blood Cancer 58 (6): 846-51, 2012.
- Montefiori LE, Bendig S, Gu Z, et al.: Enhancer Hijacking Drives Oncogenic BCL11B Expression in Lineage-Ambiguous Stem Cell Leukemia. Cancer Discov 11 (11): 2846-2867, 2021.
- Di Giacomo D, La Starza R, Gorello P, et al.: 14q32 rearrangements deregulating BCL11B mark a distinct subgroup of T-lymphoid and myeloid immature acute leukemia. Blood 138 (9): 773-784, 2021.
- Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009.
- Borowitz MJ, Béné MC, Harris NL, et al.: Acute leukaemias of ambiguous lineage. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th rev. ed. International Agency for Research on Cancer, 2017, pp 179-87.
- Davies SM, Bhatia S, Ross JA, et al.: Glutathione S-transferase genotypes, genetic susceptibility, and outcome of therapy in childhood acute lymphoblastic leukemia. Blood 100 (1): 67-71, 2002.
- Krajinovic M, Costea I, Chiasson S: Polymorphism of the thymidylate synthase gene and outcome of acute lymphoblastic leukaemia. Lancet 359 (9311): 1033-4, 2002.
- Krajinovic M, Lemieux-Blanchard E, Chiasson S, et al.: Role of polymorphisms in MTHFR and MTHFD1 genes in the outcome of childhood acute lymphoblastic leukemia. Pharmacogenomics J 4 (1): 66-72, 2004.
- Schmiegelow K, Forestier E, Kristinsson J, et al.: Thiopurine methyltransferase activity is related to the risk of relapse of childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 study. Leukemia 23 (3): 557-64, 2009.
- Relling MV, Hancock ML, Boyett JM, et al.: Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 93 (9): 2817-23, 1999.
- Stanulla M, Schaeffeler E, Flohr T, et al.: Thiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemia. JAMA 293 (12): 1485-9, 2005.
- Yang JJ, Landier W, Yang W, et al.: Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol 33 (11): 1235-42, 2015.
- Relling MV, Hancock ML, Rivera GK, et al.: Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 91 (23): 2001-8, 1999.
- Moriyama T, Nishii R, Perez-Andreu V, et al.: NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet 48 (4): 367-73, 2016.
- Tanaka Y, Kato M, Hasegawa D, et al.: Susceptibility to 6-MP toxicity conferred by a NUDT15 variant in Japanese children with acute lymphoblastic leukaemia. Br J Haematol 171 (1): 109-15, 2015.
- Diouf B, Crews KR, Lew G, et al.: Association of an inherited genetic variant with vincristine-related peripheral neuropathy in children with acute lymphoblastic leukemia. JAMA 313 (8): 815-23, 2015.
- Yang JJ, Cheng C, Yang W, et al.: Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. JAMA 301 (4): 393-403, 2009.
- Gregers J, Christensen IJ, Dalhoff K, et al.: The association of reduced folate carrier 80G>A polymorphism to outcome in childhood acute lymphoblastic leukemia interacts with chromosome 21 copy number. Blood 115 (23): 4671-7, 2010.
- Radtke S, Zolk O, Renner B, et al.: Germline genetic variations in methotrexate candidate genes are associated with pharmacokinetics, toxicity, and outcome in childhood acute lymphoblastic leukemia. Blood 121 (26): 5145-53, 2013.
- Grimwade D, Walker H, Oliver F, et al.: The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood 92 (7): 2322-33, 1998.
- Harrison CJ, Hills RK, Moorman AV, et al.: Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council Treatment trials AML 10 and 12. J Clin Oncol 28 (16): 2674-81, 2010.
- von Neuhoff C, Reinhardt D, Sander A, et al.: Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AML-BFM 98. J Clin Oncol 28 (16): 2682-9, 2010.
- Grimwade D, Hills RK, Moorman AV, et al.: Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 116 (3): 354-65, 2010.
- Creutzig U, van den Heuvel-Eibrink MM, Gibson B, et al.: Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood 120 (16): 3187-205, 2012.
- Brown P, McIntyre E, Rau R, et al.: The incidence and clinical significance of nucleophosmin mutations in childhood AML. Blood 110 (3): 979-85, 2007.
- Hollink IH, Zwaan CM, Zimmermann M, et al.: Favorable prognostic impact of NPM1 gene mutations in childhood acute myeloid leukemia, with emphasis on cytogenetically normal AML. Leukemia 23 (2): 262-70, 2009.
- Ho PA, Alonzo TA, Gerbing RB, et al.: Prevalence and prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia (AML): a report from the Children's Oncology Group. Blood 113 (26): 6558-66, 2009.
- Meshinchi S, Alonzo TA, Stirewalt DL, et al.: Clinical implications of FLT3 mutations in pediatric AML. Blood 108 (12): 3654-61, 2006.
- Tarlock K, Meshinchi S: Pediatric acute myeloid leukemia: biology and therapeutic implications of genomic variants. Pediatr Clin North Am 62 (1): 75-93, 2015.
- Bolouri H, Farrar JE, Triche T, et al.: The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med 24 (1): 103-112, 2018.
- Zarnegar-Lumley S, Alonzo TA, Gerbing RB, et al.: Characteristics and prognostic impact of IDH mutations in AML: a COG, SWOG, and ECOG analysis. Blood Adv 7 (19): 5941-5953, 2023.
- Farrar JE, Schuback HL, Ries RE, et al.: Genomic Profiling of Pediatric Acute Myeloid Leukemia Reveals a Changing Mutational Landscape from Disease Diagnosis to Relapse. Cancer Res 76 (8): 2197-205, 2016.
- Pfister SM, Reyes-Múgica M, Chan JKC, et al.: A Summary of the Inaugural WHO Classification of Pediatric Tumors: Transitioning from the Optical into the Molecular Era. Cancer Discov 12 (2): 331-355, 2022.
- Khoury JD, Solary E, Abla O, et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 36 (7): 1703-1719, 2022.
- Klein K, Kaspers G, Harrison CJ, et al.: Clinical Impact of Additional Cytogenetic Aberrations, cKIT and RAS Mutations, and Treatment Elements in Pediatric t(8;21)-AML: Results From an International Retrospective Study by the International Berlin-Frankfurt-Münster Study Group. J Clin Oncol 33 (36): 4247-58, 2015.
- Creutzig U, Zimmermann M, Ritter J, et al.: Definition of a standard-risk group in children with AML. Br J Haematol 104 (3): 630-9, 1999.
- Raimondi SC, Chang MN, Ravindranath Y, et al.: Chromosomal abnormalities in 478 children with acute myeloid leukemia: clinical characteristics and treatment outcome in a cooperative pediatric oncology group study-POG 8821. Blood 94 (11): 3707-16, 1999.
- Lie SO, Abrahamsson J, Clausen N, et al.: Treatment stratification based on initial in vivo response in acute myeloid leukaemia in children without Down's syndrome: results of NOPHO-AML trials. Br J Haematol 122 (2): 217-25, 2003.
- Duployez N, Marceau-Renaut A, Boissel N, et al.: Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood 127 (20): 2451-9, 2016.
- Faber ZJ, Chen X, Gedman AL, et al.: The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet 48 (12): 1551-1556, 2016.
- Chen W, Xie H, Wang H, et al.: Prognostic Significance of KIT Mutations in Core-Binding Factor Acute Myeloid Leukemia: A Systematic Review and Meta-Analysis. PLoS One 11 (1): e0146614, 2016.
- Shih LY, Liang DC, Huang CF, et al.: Cooperating mutations of receptor tyrosine kinases and Ras genes in childhood core-binding factor acute myeloid leukemia and a comparative analysis on paired diagnosis and relapse samples. Leukemia 22 (2): 303-7, 2008.
- Goemans BF, Zwaan CM, Miller M, et al.: Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia 19 (9): 1536-42, 2005.
- Pollard JA, Alonzo TA, Gerbing RB, et al.: Prevalence and prognostic significance of KIT mutations in pediatric patients with core binding factor AML enrolled on serial pediatric cooperative trials for de novo AML. Blood 115 (12): 2372-9, 2010.
- Shimada A, Taki T, Tabuchi K, et al.: KIT mutations, and not FLT3 internal tandem duplication, are strongly associated with a poor prognosis in pediatric acute myeloid leukemia with t(8;21): a study of the Japanese Childhood AML Cooperative Study Group. Blood 107 (5): 1806-9, 2006.
- Manara E, Bisio V, Masetti R, et al.: Core-binding factor acute myeloid leukemia in pediatric patients enrolled in the AIEOP AML 2002/01 trial: screening and prognostic impact of c-KIT mutations. Leukemia 28 (5): 1132-4, 2014.
- Chen X, Dou H, Wang X, et al.: KIT mutations correlate with adverse survival in children with core-binding factor acute myeloid leukemia. Leuk Lymphoma 59 (4): 829-836, 2018.
- Tokumasu M, Murata C, Shimada A, et al.: Adverse prognostic impact of KIT mutations in childhood CBF-AML: the results of the Japanese Pediatric Leukemia/Lymphoma Study Group AML-05 trial. Leukemia 29 (12): 2438-41, 2015.
- Tarlock K, Alonzo TA, Wang YC, et al.: Functional Properties of KIT Mutations Are Associated with Differential Clinical Outcomes and Response to Targeted Therapeutics in CBF Acute Myeloid Leukemia. Clin Cancer Res 25 (16): 5038-5048, 2019.
- Srinivasan S, Dhamne C, Patkar N, et al.: KIT exon 17 mutations are predictive of inferior outcome in pediatric acute myeloid leukemia with RUNX1::RUNX1T1. Pediatr Blood Cancer 71 (2): e30791, 2024.
- Jahn N, Agrawal M, Bullinger L, et al.: Incidence and prognostic impact of ASXL2 mutations in adult acute myeloid leukemia patients with t(8;21)(q22;q22): a study of the German-Austrian AML Study Group. Leukemia 31 (4): 1012-1015, 2017.
- Yamato G, Shiba N, Yoshida K, et al.: ASXL2 mutations are frequently found in pediatric AML patients with t(8;21)/ RUNX1-RUNX1T1 and associated with a better prognosis. Genes Chromosomes Cancer 56 (5): 382-393, 2017.
- Döhner K, Schlenk RF, Habdank M, et al.: Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood 106 (12): 3740-6, 2005.
- Verhaak RG, Goudswaard CS, van Putten W, et al.: Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood 106 (12): 3747-54, 2005.
- Schnittger S, Schoch C, Kern W, et al.: Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood 106 (12): 3733-9, 2005.
- Falini B, Mecucci C, Tiacci E, et al.: Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 352 (3): 254-66, 2005.
- Schlenk RF, Döhner K, Krauter J, et al.: Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 358 (18): 1909-18, 2008.
- Gale RE, Green C, Allen C, et al.: The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 111 (5): 2776-84, 2008.
- Cazzaniga G, Dell'Oro MG, Mecucci C, et al.: Nucleophosmin mutations in childhood acute myelogenous leukemia with normal karyotype. Blood 106 (4): 1419-22, 2005.
- Balgobind BV, Hollink IH, Arentsen-Peters ST, et al.: Integrative analysis of type-I and type-II aberrations underscores the genetic heterogeneity of pediatric acute myeloid leukemia. Haematologica 96 (10): 1478-87, 2011.
- Staffas A, Kanduri M, Hovland R, et al.: Presence of FLT3-ITD and high BAALC expression are independent prognostic markers in childhood acute myeloid leukemia. Blood 118 (22): 5905-13, 2011.
- Tawana K, Wang J, Renneville A, et al.: Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood 126 (10): 1214-23, 2015.
- Tarlock K, Lamble AJ, Wang YC, et al.: CEBPA-bZip mutations are associated with favorable prognosis in de novo AML: a report from the Children's Oncology Group. Blood 138 (13): 1137-1147, 2021.
- Marcucci G, Maharry K, Radmacher MD, et al.: Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B Study. J Clin Oncol 26 (31): 5078-87, 2008.
- Wouters BJ, Löwenberg B, Erpelinck-Verschueren CA, et al.: Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 113 (13): 3088-91, 2009.
- Dufour A, Schneider F, Metzeler KH, et al.: Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J Clin Oncol 28 (4): 570-7, 2010.
- Taskesen E, Bullinger L, Corbacioglu A, et al.: Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood 117 (8): 2469-75, 2011.
- Fasan A, Haferlach C, Alpermann T, et al.: The role of different genetic subtypes of CEBPA mutated AML. Leukemia 28 (4): 794-803, 2014.
- Wakita S, Sakaguchi M, Oh I, et al.: Prognostic impact of CEBPA bZIP domain mutation in acute myeloid leukemia. Blood Adv 6 (1): 238-247, 2022.
- Taube F, Georgi JA, Kramer M, et al.: CEBPA mutations in 4708 patients with acute myeloid leukemia: differential impact of bZIP and TAD mutations on outcome. Blood 139 (1): 87-103, 2022.
- Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, et al.: Characterization of CEBPA mutations and promoter hypermethylation in pediatric acute myeloid leukemia. Haematologica 96 (3): 384-92, 2011.
- Tarlock K, Alonzo T, Wang YC, et al.: Prognostic impact of CSF3R mutations in favorable risk childhood acute myeloid leukemia. Blood 135 (18): 1603-1606, 2020.
- Tawana K, Rio-Machin A, Preudhomme C, et al.: Familial CEBPA-mutated acute myeloid leukemia. Semin Hematol 54 (2): 87-93, 2017.
- Pui CH, Relling MV, Rivera GK, et al.: Epipodophyllotoxin-related acute myeloid leukemia: a study of 35 cases. Leukemia 9 (12): 1990-6, 1995.
- Inaba H, Zhou Y, Abla O, et al.: Heterogeneous cytogenetic subgroups and outcomes in childhood acute megakaryoblastic leukemia: a retrospective international study. Blood 126 (13): 1575-84, 2015.
- de Rooij JD, Branstetter C, Ma J, et al.: Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet 49 (3): 451-456, 2017.
- Balgobind BV, Raimondi SC, Harbott J, et al.: Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood 114 (12): 2489-96, 2009.
- Swansbury GJ, Slater R, Bain BJ, et al.: Hematological malignancies with t(9;11)(p21-22;q23)--a laboratory and clinical study of 125 cases. European 11q23 Workshop participants. Leukemia 12 (5): 792-800, 1998.
- Pollard JA, Guest E, Alonzo TA, et al.: Gemtuzumab Ozogamicin Improves Event-Free Survival and Reduces Relapse in Pediatric KMT2A-Rearranged AML: Results From the Phase III Children's Oncology Group Trial AAML0531. J Clin Oncol 39 (28): 3149-3160, 2021.
- van Weelderen RE, Klein K, Harrison CJ, et al.: Measurable Residual Disease and Fusion Partner Independently Predict Survival and Relapse Risk in Childhood KMT2A-Rearranged Acute Myeloid Leukemia: A Study by the International Berlin-Frankfurt-Münster Study Group. J Clin Oncol 41 (16): 2963-2974, 2023.
- Gröschel S, Sanders MA, Hoogenboezem R, et al.: A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157 (2): 369-81, 2014.
- Yamazaki H, Suzuki M, Otsuki A, et al.: A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 25 (4): 415-27, 2014.
- Mrózek K, Heerema NA, Bloomfield CD: Cytogenetics in acute leukemia. Blood Rev 18 (2): 115-36, 2004.
- Lugthart S, Gröschel S, Beverloo HB, et al.: Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J Clin Oncol 28 (24): 3890-8, 2010.
- Balgobind BV, Lugthart S, Hollink IH, et al.: EVI1 overexpression in distinct subtypes of pediatric acute myeloid leukemia. Leukemia 24 (5): 942-9, 2010.
- Elsherif M, Hammad M, Hafez H, et al.: MECOM gene overexpression in pediatric patients with acute myeloid leukemia. Acta Oncol 61 (4): 516-522, 2022.
- Baldazzi C, Luatti S, Zuffa E, et al.: Complex chromosomal rearrangements leading to MECOM overexpression are recurrent in myeloid malignancies with various 3q abnormalities. Genes Chromosomes Cancer 55 (4): 375-88, 2016.
- Lim G, Choi JR, Kim MJ, et al.: Detection of t(3;5) and NPM1/MLF1 rearrangement in an elderly patient with acute myeloid leukemia: clinical and laboratory study with review of the literature. Cancer Genet Cytogenet 199 (2): 101-9, 2010.
- Dumézy F, Renneville A, Mayeur-Rousse C, et al.: Acute myeloid leukemia with translocation t(3;5): new molecular insights. Haematologica 98 (4): e52-4, 2013.
- Ageberg M, Drott K, Olofsson T, et al.: Identification of a novel and myeloid specific role of the leukemia-associated fusion protein DEK-NUP214 leading to increased protein synthesis. Genes Chromosomes Cancer 47 (4): 276-87, 2008.
- Shiba N, Ichikawa H, Taki T, et al.: NUP98-NSD1 gene fusion and its related gene expression signature are strongly associated with a poor prognosis in pediatric acute myeloid leukemia. Genes Chromosomes Cancer 52 (7): 683-93, 2013.
- Slovak ML, Gundacker H, Bloomfield CD, et al.: A retrospective study of 69 patients with t(6;9)(p23;q34) AML emphasizes the need for a prospective, multicenter initiative for rare 'poor prognosis' myeloid malignancies. Leukemia 20 (7): 1295-7, 2006.
- Alsabeh R, Brynes RK, Slovak ML, et al.: Acute myeloid leukemia with t(6;9) (p23;q34): association with myelodysplasia, basophilia, and initial CD34 negative immunophenotype. Am J Clin Pathol 107 (4): 430-7, 1997.
- Sandahl JD, Coenen EA, Forestier E, et al.: t(6;9)(p22;q34)/DEK-NUP214-rearranged pediatric myeloid leukemia: an international study of 62 patients. Haematologica 99 (5): 865-72, 2014.
- Tarlock K, Alonzo TA, Moraleda PP, et al.: Acute myeloid leukaemia (AML) with t(6;9)(p23;q34) is associated with poor outcome in childhood AML regardless of FLT3-ITD status: a report from the Children's Oncology Group. Br J Haematol 166 (2): 254-9, 2014.
- Lamble AJ, Hagiwara K, Gerbing RB, et al.: CREBBP alterations are associated with a poor prognosis in de novo AML. Blood 141 (17): 2156-2159, 2023.
- Coenen EA, Zwaan CM, Reinhardt D, et al.: Pediatric acute myeloid leukemia with t(8;16)(p11;p13), a distinct clinical and biological entity: a collaborative study by the International-Berlin-Frankfurt-Munster AML-study group. Blood 122 (15): 2704-13, 2013.
- Wong KF, Yuen HL, Siu LL, et al.: t(8;16)(p11;p13) predisposes to a transient but potentially recurring neonatal leukemia. Hum Pathol 39 (11): 1702-7, 2008.
- Wu X, Sulavik D, Roulston D, et al.: Spontaneous remission of congenital acute myeloid leukemia with t(8;16)(p11;13). Pediatr Blood Cancer 56 (2): 331-2, 2011.
- Terui K, Sato T, Sasaki S, et al.: Two novel variants of MOZ-CBP fusion transcripts in spontaneously remitted infant leukemia with t(1;16;8)(p13;p13;p11), a new variant of t(8;16)(p11;p13). Haematologica 93 (10): 1591-3, 2008.
- Noort S, Zimmermann M, Reinhardt D, et al.: Prognostic impact of t(16;21)(p11;q22) and t(16;21)(q24;q22) in pediatric AML: a retrospective study by the I-BFM Study Group. Blood 132 (15): 1584-1592, 2018.
- Gruber TA, Larson Gedman A, Zhang J, et al.: An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell 22 (5): 683-97, 2012.
- Thiollier C, Lopez CK, Gerby B, et al.: Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J Exp Med 209 (11): 2017-31, 2012.
- de Rooij JD, Hollink IH, Arentsen-Peters ST, et al.: NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia 27 (12): 2280-8, 2013.
- Masetti R, Pigazzi M, Togni M, et al.: CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood 121 (17): 3469-72, 2013.
- Masetti R, Rondelli R, Fagioli F, et al.: Infants with acute myeloid leukemia treated according to the Associazione Italiana di Ematologia e Oncologia Pediatrica 2002/01 protocol have an outcome comparable to that of older children. Haematologica 99 (8): e127-9, 2014.
- de Rooij JD, Masetti R, van den Heuvel-Eibrink MM, et al.: Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: a retrospective intergroup study. Blood 127 (26): 3424-30, 2016.
- Hara Y, Shiba N, Ohki K, et al.: Prognostic impact of specific molecular profiles in pediatric acute megakaryoblastic leukemia in non-Down syndrome. Genes Chromosomes Cancer 56 (5): 394-404, 2017.
- Chisholm KM, Smith J, Heerema-McKenney AE, et al.: Pathologic, cytogenetic, and molecular features of acute myeloid leukemia with megakaryocytic differentiation: A report from the Children's Oncology Group. Pediatr Blood Cancer 70 (5): e30251, 2023.
- Eidenschink Brodersen L, Alonzo TA, Menssen AJ, et al.: A recurrent immunophenotype at diagnosis independently identifies high-risk pediatric acute myeloid leukemia: a report from Children's Oncology Group. Leukemia 30 (10): 2077-2080, 2016.
- Pardo LM, Voigt AP, Alonzo TA, et al.: Deciphering the Significance of CD56 Expression in Pediatric Acute Myeloid Leukemia: A Report from the Children's Oncology Group. Cytometry B Clin Cytom 98 (1): 52-56, 2020.
- Smith JL, Ries RE, Hylkema T, et al.: Comprehensive Transcriptome Profiling of Cryptic CBFA2T3-GLIS2 Fusion-Positive AML Defines Novel Therapeutic Options: A COG and TARGET Pediatric AML Study. Clin Cancer Res 26 (3): 726-737, 2020.
- Tang T, Le Q, Castro S, et al.: Targeting FOLR1 in high-risk CBF2AT3-GLIS2 pediatric AML with STRO-002 FOLR1-antibody-drug conjugate. Blood Adv 6 (22): 5933-5937, 2022.
- Le Q, Hadland B, Smith JL, et al.: CBFA2T3-GLIS2 model of pediatric acute megakaryoblastic leukemia identifies FOLR1 as a CAR T cell target. J Clin Invest 132 (22): , 2022.
- Takeda A, Yaseen NR: Nucleoporins and nucleocytoplasmic transport in hematologic malignancies. Semin Cancer Biol 27: 3-10, 2014.
- Bertrums EJM, Smith JL, Harmon L, et al.: Comprehensive molecular and clinical characterization of NUP98 fusions in pediatric acute myeloid leukemia. Haematologica 108 (8): 2044-2058, 2023.
- Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, et al.: NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood 118 (13): 3645-56, 2011.
- Ostronoff F, Othus M, Gerbing RB, et al.: NUP98/NSD1 and FLT3/ITD coexpression is more prevalent in younger AML patients and leads to induction failure: a COG and SWOG report. Blood 124 (15): 2400-7, 2014.
- Panarello C, Rosanda C, Morerio C: Cryptic translocation t(5;11)(q35;p15.5) with involvement of the NSD1 and NUP98 genes without 5q deletion in childhood acute myeloid leukemia. Genes Chromosomes Cancer 35 (3): 277-81, 2002.
- Struski S, Lagarde S, Bories P, et al.: NUP98 is rearranged in 3.8% of pediatric AML forming a clinical and molecular homogenous group with a poor prognosis. Leukemia 31 (3): 565-572, 2017.
- Cerveira N, Correia C, Dória S, et al.: Frequency of NUP98-NSD1 fusion transcript in childhood acute myeloid leukaemia. Leukemia 17 (11): 2244-7, 2003.
- McNeer NA, Philip J, Geiger H, et al.: Genetic mechanisms of primary chemotherapy resistance in pediatric acute myeloid leukemia. Leukemia 33 (8): 1934-1943, 2019.
- van Zutven LJ, Onen E, Velthuizen SC, et al.: Identification of NUP98 abnormalities in acute leukemia: JARID1A (12p13) as a new partner gene. Genes Chromosomes Cancer 45 (5): 437-46, 2006.
- Noort S, Wander P, Alonzo TA, et al.: The clinical and biological characteristics of NUP98-KDM5A in pediatric acute myeloid leukemia. Haematologica 106 (2): 630-634, 2021.
- Espersen ADL, Noren-Nyström U, Abrahamsson J, et al.: Acute myeloid leukemia (AML) with t(7;12)(q36;p13) is associated with infancy and trisomy 19: Data from Nordic Society for Pediatric Hematology and Oncology (NOPHO-AML) and review of the literature. Genes Chromosomes Cancer 57 (7): 359-365, 2018.
- von Bergh AR, van Drunen E, van Wering ER, et al.: High incidence of t(7;12)(q36;p13) in infant AML but not in infant ALL, with a dismal outcome and ectopic expression of HLXB9. Genes Chromosomes Cancer 45 (8): 731-9, 2006.
- Tosi S, Harbott J, Teigler-Schlegel A, et al.: t(7;12)(q36;p13), a new recurrent translocation involving ETV6 in infant leukemia. Genes Chromosomes Cancer 29 (4): 325-32, 2000.
- Slater RM, von Drunen E, Kroes WG, et al.: t(7;12)(q36;p13) and t(7;12)(q32;p13)--translocations involving ETV6 in children 18 months of age or younger with myeloid disorders. Leukemia 15 (6): 915-20, 2001.
- Park J, Kim M, Lim J, et al.: Three-way complex translocations in infant acute myeloid leukemia with t(7;12)(q36;p13): the incidence and correlation of a HLXB9 overexpression. Cancer Genet Cytogenet 191 (2): 102-5, 2009.
- de Rooij J, Beuling E, Fornerod M, et al.: ETV6 Aberrations Are a Recurrent Event in Pediatric Acute Myeloid Leukemia with Poor Clinical Outcome. [Abstract] Blood 124 (21): 1012, 2014. Also available online. Last accessed January 25, 2024.
- Helton HL, Ries RE, Alonzo TA, et al.: Clinically Significant Mutations, Deletions and Translocations Involving ETV6 Identified by Whole Genome and Whole Exome Sequencing; Report From NCI/COG Target AML Initiative. [Abstract] Blood 120 (21): 125, 2012. Also available online. Last accessed January 25, 2024.
- Papadopoulou V, Schoumans J, Scarpelli I, et al.: Description of an Institutional Cohort of Myeloid Neoplasms Carrying ETV6-Locus Deletions or ETV6 Rearrangements. Acta Haematol 146 (5): 401-407, 2023.
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Outcome of pediatric patients with acute myeloid leukemia (AML) and -5/5q- abnormalities from five pediatric AML treatment protocols: a report from the Children's Oncology Group. Pediatr Blood Cancer 60 (12): 2073-8, 2013.
- Stevens RF, Hann IM, Wheatley K, et al.: Marked improvements in outcome with chemotherapy alone in paediatric acute myeloid leukemia: results of the United Kingdom Medical Research Council's 10th AML trial. MRC Childhood Leukaemia Working Party. Br J Haematol 101 (1): 130-40, 1998.
- Hasle H, Alonzo TA, Auvrignon A, et al.: Monosomy 7 and deletion 7q in children and adolescents with acute myeloid leukemia: an international retrospective study. Blood 109 (11): 4641-7, 2007.
- Rasche M, von Neuhoff C, Dworzak M, et al.: Genotype-outcome correlations in pediatric AML: the impact of a monosomal karyotype in trial AML-BFM 2004. Leukemia 31 (12): 2807-2814, 2017.
- Blink M, Zimmermann M, von Neuhoff C, et al.: Normal karyotype is a poor prognostic factor in myeloid leukemia of Down syndrome: a retrospective, international study. Haematologica 99 (2): 299-307, 2014.
- Abla O, Ries RE, Triche T, et al.: Structural variants involving MLLT10 fusion are associated with adverse outcomes in pediatric acute myeloid leukemia. Blood Adv 8 (8): 2005-2017, 2024.
- Mark C, Meshinchi S, Joyce B, et al.: Treatment outcomes of childhood PICALM::MLLT10 acute leukaemias. Br J Haematol 204 (2): 576-584, 2024.
- Schnittger S, Schoch C, Dugas M, et al.: Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 100 (1): 59-66, 2002.
- Thiede C, Steudel C, Mohr B, et al.: Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 99 (12): 4326-35, 2002.
- Iwai T, Yokota S, Nakao M, et al.: Internal tandem duplication of the FLT3 gene and clinical evaluation in childhood acute myeloid leukemia. The Children's Cancer and Leukemia Study Group, Japan. Leukemia 13 (1): 38-43, 1999.
- Meshinchi S, Stirewalt DL, Alonzo TA, et al.: Activating mutations of RTK/ras signal transduction pathway in pediatric acute myeloid leukemia. Blood 102 (4): 1474-9, 2003.
- Zwaan CM, Meshinchi S, Radich JP, et al.: FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: prognostic significance and relation to cellular drug resistance. Blood 102 (7): 2387-94, 2003.
- Voigt AP, Brodersen LE, Alonzo TA, et al.: Phenotype in combination with genotype improves outcome prediction in acute myeloid leukemia: a report from Children's Oncology Group protocol AAML0531. Haematologica 102 (12): 2058-2068, 2017.
- Berman JN, Gerbing RB, Alonzo TA, et al.: Prevalence and clinical implications of NRAS mutations in childhood AML: a report from the Children's Oncology Group. Leukemia 25 (6): 1039-42, 2011.
- Kühn MW, Radtke I, Bullinger L, et al.: High-resolution genomic profiling of adult and pediatric core-binding factor acute myeloid leukemia reveals new recurrent genomic alterations. Blood 119 (10): e67-75, 2012.
- Lion T, Haas OA: Acute megakaryocytic leukemia with the t(1;22)(p13;q13). Leuk Lymphoma 11 (1-2): 15-20, 1993.
- Duchayne E, Fenneteau O, Pages MP, et al.: Acute megakaryoblastic leukaemia: a national clinical and biological study of 53 adult and childhood cases by the Groupe Français d'Hématologie Cellulaire (GFHC). Leuk Lymphoma 44 (1): 49-58, 2003.
- Ma Z, Morris SW, Valentine V, et al.: Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat Genet 28 (3): 220-1, 2001.
- Mercher T, Coniat MB, Monni R, et al.: Involvement of a human gene related to the Drosophila spen gene in the recurrent t(1;22) translocation of acute megakaryocytic leukemia. Proc Natl Acad Sci U S A 98 (10): 5776-9, 2001.
- Bernstein J, Dastugue N, Haas OA, et al.: Nineteen cases of the t(1;22)(p13;q13) acute megakaryblastic leukaemia of infants/children and a review of 39 cases: report from a t(1;22) study group. Leukemia 14 (1): 216-8, 2000.
- Hammer ASB, Juul-Dam KL, Sandahl JD, et al.: Hypodiploidy has unfavorable impact on survival in pediatric acute myeloid leukemia: an I-BFM Study Group collaboration. Blood Adv 7 (6): 1045-1055, 2023.
- Umeda M, Ma J, Huang BJ, et al.: Integrated Genomic Analysis Identifies UBTF Tandem Duplications as a Recurrent Lesion in Pediatric Acute Myeloid Leukemia. Blood Cancer Discov 3 (3): 194-207, 2022.
- Ryland GL, Umeda M, Holmfeldt L, et al.: Description of a novel subtype of acute myeloid leukemia defined by recurrent CBFB insertions. Blood 141 (7): 800-805, 2023.
- Rungjirajittranon T, Siriwannangkul T, Kungwankiattichai S, et al.: Clinical Outcomes of Acute Myeloid Leukemia Patients Harboring the RUNX1 Mutation: Is It Still an Unfavorable Prognosis? A Cohort Study and Meta-Analysis. Cancers (Basel) 14 (21): , 2022.
- Yamato G, Shiba N, Yoshida K, et al.: RUNX1 mutations in pediatric acute myeloid leukemia are associated with distinct genetic features and an inferior prognosis. Blood 131 (20): 2266-2270, 2018.
- Sendker S, Awada A, Domagalla S, et al.: RUNX1 mutation has no prognostic significance in paediatric AML: a retrospective study of the AML-BFM study group. Leukemia 37 (7): 1435-1443, 2023.
- Paschka P, Marcucci G, Ruppert AS, et al.: Wilms' tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol 26 (28): 4595-602, 2008.
- Virappane P, Gale R, Hills R, et al.: Mutation of the Wilms' tumor 1 gene is a poor prognostic factor associated with chemotherapy resistance in normal karyotype acute myeloid leukemia: the United Kingdom Medical Research Council Adult Leukaemia Working Party. J Clin Oncol 26 (33): 5429-35, 2008.
- Gaidzik VI, Schlenk RF, Moschny S, et al.: Prognostic impact of WT1 mutations in cytogenetically normal acute myeloid leukemia: a study of the German-Austrian AML Study Group. Blood 113 (19): 4505-11, 2009.
- Renneville A, Boissel N, Zurawski V, et al.: Wilms tumor 1 gene mutations are associated with a higher risk of recurrence in young adults with acute myeloid leukemia: a study from the Acute Leukemia French Association. Cancer 115 (16): 3719-27, 2009.
- Ho PA, Zeng R, Alonzo TA, et al.: Prevalence and prognostic implications of WT1 mutations in pediatric acute myeloid leukemia (AML): a report from the Children's Oncology Group. Blood 116 (5): 702-10, 2010.
- Hollink IH, van den Heuvel-Eibrink MM, Zimmermann M, et al.: Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood 113 (23): 5951-60, 2009.
- Ley TJ, Ding L, Walter MJ, et al.: DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363 (25): 2424-33, 2010.
- Yan XJ, Xu J, Gu ZH, et al.: Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet 43 (4): 309-15, 2011.
- Thol F, Damm F, Lüdeking A, et al.: Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol 29 (21): 2889-96, 2011.
- Ho PA, Kutny MA, Alonzo TA, et al.: Leukemic mutations in the methylation-associated genes DNMT3A and IDH2 are rare events in pediatric AML: a report from the Children's Oncology Group. Pediatr Blood Cancer 57 (2): 204-9, 2011.
- Green CL, Evans CM, Hills RK, et al.: The prognostic significance of IDH1 mutations in younger adult patients with acute myeloid leukemia is dependent on FLT3/ITD status. Blood 116 (15): 2779-82, 2010.
- Paschka P, Schlenk RF, Gaidzik VI, et al.: IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol 28 (22): 3636-43, 2010.
- Abbas S, Lugthart S, Kavelaars FG, et al.: Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood 116 (12): 2122-6, 2010.
- Marcucci G, Maharry K, Wu YZ, et al.: IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 28 (14): 2348-55, 2010.
- Wagner K, Damm F, Göhring G, et al.: Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J Clin Oncol 28 (14): 2356-64, 2010.
- Figueroa ME, Abdel-Wahab O, Lu C, et al.: Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18 (6): 553-67, 2010.
- Ward PS, Patel J, Wise DR, et al.: The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17 (3): 225-34, 2010.
- Dang L, White DW, Gross S, et al.: Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462 (7274): 739-44, 2009.
- Damm F, Thol F, Hollink I, et al.: Prevalence and prognostic value of IDH1 and IDH2 mutations in childhood AML: a study of the AML-BFM and DCOG study groups. Leukemia 25 (11): 1704-10, 2011.
- Oki K, Takita J, Hiwatari M, et al.: IDH1 and IDH2 mutations are rare in pediatric myeloid malignancies. Leukemia 25 (2): 382-4, 2011.
- Pigazzi M, Ferrari G, Masetti R, et al.: Low prevalence of IDH1 gene mutation in childhood AML in Italy. Leukemia 25 (1): 173-4, 2011.
- Ho PA, Alonzo TA, Kopecky KJ, et al.: Molecular alterations of the IDH1 gene in AML: a Children's Oncology Group and Southwest Oncology Group study. Leukemia 24 (5): 909-13, 2010.
- Andersson AK, Miller DW, Lynch JA, et al.: IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia 25 (10): 1570-7, 2011.
- Maxson JE, Ries RE, Wang YC, et al.: CSF3R mutations have a high degree of overlap with CEBPA mutations in pediatric AML. Blood 127 (24): 3094-8, 2016.
- Germeshausen M, Kratz CP, Ballmaier M, et al.: RAS and CSF3R mutations in severe congenital neutropenia. Blood 114 (16): 3504-5, 2009.
- Skokowa J, Steinemann D, Katsman-Kuipers JE, et al.: Cooperativity of RUNX1 and CSF3R mutations in severe congenital neutropenia: a unique pathway in myeloid leukemogenesis. Blood 123 (14): 2229-37, 2014.
- Melnick A, Licht JD: Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 93 (10): 3167-215, 1999.
- Sanz MA, Grimwade D, Tallman MS, et al.: Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 113 (9): 1875-91, 2009.
- Falini B, Flenghi L, Fagioli M, et al.: Immunocytochemical diagnosis of acute promyelocytic leukemia (M3) with the monoclonal antibody PG-M3 (anti-PML). Blood 90 (10): 4046-53, 1997.
- Gomis F, Sanz J, Sempere A, et al.: Immunofluorescent analysis with the anti-PML monoclonal antibody PG-M3 for rapid and accurate genetic diagnosis of acute promyelocytic leukemia. Ann Hematol 83 (11): 687-90, 2004.
- Dimov ND, Medeiros LJ, Kantarjian HM, et al.: Rapid and reliable confirmation of acute promyelocytic leukemia by immunofluorescence staining with an antipromyelocytic leukemia antibody: the M. D. Anderson Cancer Center experience of 349 patients. Cancer 116 (2): 369-76, 2010.
- Zelent A, Guidez F, Melnick A, et al.: Translocations of the RARalpha gene in acute promyelocytic leukemia. Oncogene 20 (49): 7186-203, 2001.
- Yan W, Zhang G: Molecular Characteristics and Clinical Significance of 12 Fusion Genes in Acute Promyelocytic Leukemia: A Systematic Review. Acta Haematol 136 (1): 1-15, 2016.
- Rego EM, Ruggero D, Tribioli C, et al.: Leukemia with distinct phenotypes in transgenic mice expressing PML/RAR alpha, PLZF/RAR alpha or NPM/RAR alpha. Oncogene 25 (13): 1974-9, 2006.
- Licht JD, Chomienne C, Goy A, et al.: Clinical and molecular characterization of a rare syndrome of acute promyelocytic leukemia associated with translocation (11;17). Blood 85 (4): 1083-94, 1995.
- Guidez F, Ivins S, Zhu J, et al.: Reduced retinoic acid-sensitivities of nuclear receptor corepressor binding to PML- and PLZF-RARalpha underlie molecular pathogenesis and treatment of acute promyelocytic leukemia. Blood 91 (8): 2634-42, 1998.
- Grimwade D, Biondi A, Mozziconacci MJ, et al.: Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Groupe Français de Cytogénétique Hématologique, Groupe de Français d'Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action "Molecular Cytogenetic Diagnosis in Haematological Malignancies". Blood 96 (4): 1297-308, 2000.
- Sukhai MA, Wu X, Xuan Y, et al.: Myeloid leukemia with promyelocytic features in transgenic mice expressing hCG-NuMA-RARalpha. Oncogene 23 (3): 665-78, 2004.
- Redner RL, Corey SJ, Rush EA: Differentiation of t(5;17) variant acute promyelocytic leukemic blasts by all-trans retinoic acid. Leukemia 11 (7): 1014-6, 1997.
- Wells RA, Catzavelos C, Kamel-Reid S: Fusion of retinoic acid receptor alpha to NuMA, the nuclear mitotic apparatus protein, by a variant translocation in acute promyelocytic leukaemia. Nat Genet 17 (1): 109-13, 1997.
- Wells RA, Hummel JL, De Koven A, et al.: A new variant translocation in acute promyelocytic leukaemia: molecular characterization and clinical correlation. Leukemia 10 (4): 735-40, 1996.
- Chen X, Wang F, Zhou X, et al.: Torque teno mini virus driven childhood acute promyelocytic leukemia: The third case report and sequence analysis. Front Oncol 12: 1074913, 2022.
- Sala-Torra O, Beppu LW, Abukar FA, et al.: TTMV-RARA fusion as a recurrent cause of AML with APL characteristics. Blood Adv 6 (12): 3590-3592, 2022.
- Callens C, Chevret S, Cayuela JM, et al.: Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia 19 (7): 1153-60, 2005.
- Gale RE, Hills R, Pizzey AR, et al.: Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood 106 (12): 3768-76, 2005.
- Arrigoni P, Beretta C, Silvestri D, et al.: FLT3 internal tandem duplication in childhood acute myeloid leukaemia: association with hyperleucocytosis in acute promyelocytic leukaemia. Br J Haematol 120 (1): 89-92, 2003.
- Noguera NI, Breccia M, Divona M, et al.: Alterations of the FLT3 gene in acute promyelocytic leukemia: association with diagnostic characteristics and analysis of clinical outcome in patients treated with the Italian AIDA protocol. Leukemia 16 (11): 2185-9, 2002.
- Tallman MS, Kim HT, Montesinos P, et al.: Does microgranular variant morphology of acute promyelocytic leukemia independently predict a less favorable outcome compared with classical M3 APL? A joint study of the North American Intergroup and the PETHEMA Group. Blood 116 (25): 5650-9, 2010.
- Iland HJ, Bradstock K, Supple SG, et al.: All-trans-retinoic acid, idarubicin, and IV arsenic trioxide as initial therapy in acute promyelocytic leukemia (APML4). Blood 120 (8): 1570-80; quiz 1752, 2012.
- Kutny MA, Moser BK, Laumann K, et al.: FLT3 mutation status is a predictor of early death in pediatric acute promyelocytic leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 59 (4): 662-7, 2012.
- Kutny MA, Alonzo TA, Abla O, et al.: Assessment of Arsenic Trioxide and All-trans Retinoic Acid for the Treatment of Pediatric Acute Promyelocytic Leukemia: A Report From the Children's Oncology Group AAML1331 Trial. JAMA Oncol 8 (1): 79-87, 2022.
- Quintás-Cardama A, Cortes J: Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 113 (8): 1619-30, 2009.
- Loghavi S, Kanagal-Shamanna R, Khoury JD, et al.: Fifth Edition of the World Health Classification of Tumors of the Hematopoietic and Lymphoid Tissue: Myeloid Neoplasms. Mod Pathol 37 (2): 100397, 2024.
- Wang W, Cortes JE, Tang G, et al.: Risk stratification of chromosomal abnormalities in chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Blood 127 (22): 2742-50, 2016.
- Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47 (11): 1334-40, 2015.
- Stieglitz E, Taylor-Weiner AN, Chang TY, et al.: The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 47 (11): 1326-33, 2015.
- Murakami N, Okuno Y, Yoshida K, et al.: Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood 131 (14): 1576-1586, 2018.
- Sakaguchi H, Okuno Y, Muramatsu H, et al.: Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet 45 (8): 937-41, 2013.
- Stieglitz E, Mazor T, Olshen AB, et al.: Genome-wide DNA methylation is predictive of outcome in juvenile myelomonocytic leukemia. Nat Commun 8 (1): 2127, 2017.
- Hecht A, Meyer JA, Behnert A, et al.: Molecular and phenotypic diversity of CBL-mutated juvenile myelomonocytic leukemia. Haematologica 107 (1): 178-186, 2022.
- Helsmoortel HH, Bresolin S, Lammens T, et al.: LIN28B overexpression defines a novel fetal-like subgroup of juvenile myelomonocytic leukemia. Blood 127 (9): 1163-72, 2016.
- Schwartz JR, Ma J, Lamprecht T, et al.: The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun 8 (1): 1557, 2017.
- Pastor V, Hirabayashi S, Karow A, et al.: Mutational landscape in children with myelodysplastic syndromes is distinct from adults: specific somatic drivers and novel germline variants. Leukemia 31 (3): 759-762, 2017.
- Collin M, Dickinson R, Bigley V: Haematopoietic and immune defects associated with GATA2 mutation. Br J Haematol 169 (2): 173-87, 2015.
- Wlodarski MW, Hirabayashi S, Pastor V, et al.: Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 127 (11): 1387-97; quiz 1518, 2016.
- Wlodarski MW, Collin M, Horwitz MS: GATA2 deficiency and related myeloid neoplasms. Semin Hematol 54 (2): 81-86, 2017.
- Davidsson J, Puschmann A, Tedgård U, et al.: SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 32 (5): 1106-1115, 2018.
- Schwartz JR, Wang S, Ma J, et al.: Germline SAMD9 mutation in siblings with monosomy 7 and myelodysplastic syndrome. Leukemia 31 (8): 1827-1830, 2017.
- Narumi S, Amano N, Ishii T, et al.: SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet 48 (7): 792-7, 2016.
- Chen DH, Below JE, Shimamura A, et al.: Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am J Hum Genet 98 (6): 1146-1158, 2016.
- Wong JC, Bryant V, Lamprecht T, et al.: Germline SAMD9 and SAMD9L mutations are associated with extensive genetic evolution and diverse hematologic outcomes. JCI Insight 3 (14): , 2018.
- Göhring G, Michalova K, Beverloo HB, et al.: Complex karyotype newly defined: the strongest prognostic factor in advanced childhood myelodysplastic syndrome. Blood 116 (19): 3766-9, 2010.
- Haase D, Germing U, Schanz J, et al.: New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood 110 (13): 4385-95, 2007.
- Arber DA, Vardiman JW, Brunning RD: Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 110-23.
- Hitzler JK, Cheung J, Li Y, et al.: GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood 101 (11): 4301-4, 2003.
- Mundschau G, Gurbuxani S, Gamis AS, et al.: Mutagenesis of GATA1 is an initiating event in Down syndrome leukemogenesis. Blood 101 (11): 4298-300, 2003.
- Massey GV, Zipursky A, Chang MN, et al.: A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children's Oncology Group (COG) study POG-9481. Blood 107 (12): 4606-13, 2006.
- Groet J, McElwaine S, Spinelli M, et al.: Acquired mutations in GATA1 in neonates with Down's syndrome with transient myeloid disorder. Lancet 361 (9369): 1617-20, 2003.
- Rainis L, Bercovich D, Strehl S, et al.: Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood 102 (3): 981-6, 2003.
- Wechsler J, Greene M, McDevitt MA, et al.: Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet 32 (1): 148-52, 2002.
- Ge Y, Stout ML, Tatman DA, et al.: GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst 97 (3): 226-31, 2005.
Non-Hodgkin Lymphoma
Mature B-cell Lymphoma
The mature B-cell lymphomas include Burkitt lymphoma, diffuse large B-cell lymphoma, and primary mediastinal B-cell lymphoma.
Burkitt lymphoma
Genomics of Burkitt lymphoma
The malignant cells of Burkitt lymphoma show a mature B-cell phenotype and are negative for the enzyme terminal deoxynucleotidyl transferase. These malignant cells usually express surface immunoglobulin (Ig), most bearing a clonal surface IgM with either kappa or lambda light chains. A variety of additional B-cell markers (e.g., CD19, CD20, CD22) are usually present, and most childhood Burkitt lymphomas express CD10.
Burkitt lymphoma expresses a characteristic chromosomal translocation, usually t(8;14) and more rarely t(8;22) or t(2;8). Each of these translocations juxtaposes the MYC oncogene and the immunoglobulin locus (IG, mostly the IGH locus) regulatory elements, resulting in the inappropriate expression of MYC, a gene involved in cellular proliferation. The presence of one of the variant translocations t(2;8) or t(8;22) does not appear to affect response or outcome.
Mapping of IGH-translocation breakpoints demonstrated that IG::MYC translocations in sporadic Burkitt lymphoma most commonly occur through aberrant class-switch recombination and less commonly through somatic hypervariant. Translocations resulting from aberrant variable, diversity, and joining (VDJ) gene segment recombinations are rare. These findings are consistent with a germinal center derivation of Burkitt lymphoma.
While MYC translocations are present in all Burkitt lymphoma, cooperating genomic alterations appear to be required for lymphoma development. Some of the more commonly observed recurring variants that have been identified in Burkitt lymphoma in pediatric and adult cases are listed below. The clinical significance of these variants for pediatric Burkitt lymphoma remains to be elucidated.
- Activating variants in the transcription factor TCF3 and inactivating variants in its negative regulator ID3 are observed in approximately 70% of Burkitt lymphoma cases.
- TP53 variants are observed in one-third to one-half of cases.
- CCND3 variants are commonly observed in sporadic Burkitt lymphoma (approximately 40% of cases) but are rare in endemic Burkitt lymphoma.
- Mutually exclusive variants in SMARCA4 and ARID1A, components of the SWItch/Sucrose Non-Fermentable (SWI/SNF) complex, are observed in more than one-half of pediatric Burkitt lymphoma cases.
- Variants in MYC itself are observed in approximately one-half of Burkitt lymphoma cases and appear to enhance tumorigenesis, in part, by increasing MYC stability.
- Variants and altered DNA methylation result in dysregulation of sphingosine-1-phosphate signaling in a subset of Burkitt lymphoma. Genes contributing to this include RHOA, which is altered in approximately 10% of cases, and, less commonly, GNA13, GNA11, and GNA12.
A study that compared the genomic landscape of endemic Burkitt lymphoma with the genomics of sporadic Burkitt lymphoma found the expected high rate of Epstein-Barr virus (EBV) positivity in endemic cases, with much lower rates in sporadic cases. There was general similarity between the patterns of variants for endemic and sporadic cases and for EBV-positive and EBV-negative cases. However, EBV-positive cases showed significantly lower variant rates for selected genes/pathways, including SMARCA4, CCND3, TP53, and apoptosis.
Cytogenetic evidence of MYC rearrangement is the gold standard for diagnosis of Burkitt lymphoma. For cases in which cytogenetic analysis is not available, the World Health Organization (WHO) has recommended that the Burkitt-like diagnosis be reserved for lymphoma resembling Burkitt lymphoma or with more pleomorphism, large cells, and a proliferation fraction (i.e., MIB-1 or Ki-67 immunostaining) of 99% or greater. BCL2 staining by immunohistochemistry is variable. The absence of a translocation involving the BCL2 gene does not preclude the diagnosis of Burkitt lymphoma and has no clinical implications.
Genomics of Burkitt-like lymphoma/high-grade B-cell lymphoma with 11q aberrations
Burkitt-like lymphoma with 11q aberration was added as a provisional entity in the 2017 revised WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. In the 5th edition of the WHO classification, this entity was renamed high-grade B-cell lymphoma with 11q aberrations. In this entity, MYC rearrangement is absent, and the characteristic chromosome 11q finding (detected cytogenetically and/or with copy-number DNA arrays) is 11q23.2-q23.3 gain/amplification and 11q24.1-qter loss.
- In a study of 102 lymphomas that morphologically resembled Burkitt lymphoma, diffuse large B-cell lymphoma, and high-grade B-cell lymphoma, unclassifiable, 13 cases (13%) lacked a MYC rearrangement but were positive for 11q proximal gain and telomeric loss by fluorescence in situ hybridization.
- Most patients with high-grade B-cell lymphoma with 11q aberrations present in the adolescent and young adult age range with localized nodal disease. Head and neck involvement is the most common presentation, although presentation in other nodal areas, as well as in the abdomen, can occur.
- Cases show a very high proliferative index and can show a focal starry sky pattern.
- Outcomes appear highly favorable in the small number of cases identified.
- The variant landscape of high-grade B-cell lymphoma with 11q aberrations is distinct from that of Burkitt lymphoma. Variants commonly observed in Burkitt lymphoma (e.g., ID3, TCF3, and CCND3) are uncommon in high-grade B-cell lymphoma with 11q aberrations. Conversely, variants in GNA13 appear to be common (up to 50%) in patients with high-grade B-cell lymphoma with 11q aberrations and are less common in patients with Burkitt lymphoma.
For information about the treatment of childhood Burkitt lymphoma, see Childhood Non-Hodgkin Lymphoma Treatment.
Diffuse large B-cell lymphoma
Genomics of diffuse large B-cell lymphoma
Gene expression profiling of diffuse large B-cell lymphoma in adults has defined molecular subtypes. These subtypes are based on the suspected cell of origin, including germinal center B cell (GCB), activated B cell (ABC), and 10% to 15% of cases that remain unclassifiable. Current comprehensive molecular profiling of diffuse large B-cell lymphoma in adults has led to the proposal of additional subclassification beyond the cell of origin. This additional subclassification is based on genetic variants and copy number variations. Diffuse large B-cell lymphoma in children and adolescents differs biologically from diffuse large B-cell lymphoma in adults in the following ways:
- Most pediatric diffuse large B-cell lymphoma cases have a germinal center B-cell phenotype, as assessed by immunohistochemical analysis of selected proteins found in normal germinal center B cells, such as the BCL6 gene product and CD10. The age at which the favorable germinal center subtype changes to the less favorable nongerminal center subtype was shown to be a continuous variable.
- Pediatric diffuse large B-cell lymphoma rarely demonstrates the t(14;18) translocation involving the IGH gene and the BCL2 gene that is seen in adults.
- As many as 30% of patients younger than 14 years with diffuse large B-cell lymphoma will have a gene signature similar to Burkitt lymphoma.
- In contrast to adult diffuse large B-cell lymphoma, pediatric cases show a high frequency of abnormalities at the MYC locus (chromosome 8q24), with approximately one-third of pediatric cases showing MYC rearrangement and approximately one-half of the nonrearranged cases showing MYC gain or amplification.
- One report included 31 pediatric patients with diffuse large B-cell lymphoma, NOS. Most patients (n = 21) showed a germinal center phenotype, and the genomic alterations resembled those of adult germinal center B-cell diffuse large B-cell lymphoma (GCB-DLBCL) (e.g., SOCS1 and KMT2D variants). Among this group of patients, MYC rearrangements were detected in 3 patients, and 5 of 25 cases were EBV positive (4 with the activated B-cell phenotype).
Large B-cell lymphoma with IRF4 rearrangement (LBCL-IRF4) is a distinct entity in the 5th edition of the WHO classification of lymphoid neoplasms.
- LBCL-IRF4 cases have a translocation that juxtaposes the IRF4 oncogene next to one of the immunoglobulin loci.
- In one report, diffuse large B-cell lymphoma cases with an IRF4 translocation were significantly more frequent in children than in adults with diffuse large B-cell lymphoma or follicular lymphoma (15% vs. 2%). One study of 32 pediatric cases of diffuse large B-cell lymphoma or follicular lymphoma found 2 (6%) with IRF4 translocations. A second study of 34 cases of pediatric follicular lymphoma or diffuse large B-cell lymphoma found 7 cases (21%) with IRF translocations. Most of these cases occurred in the adolescent age range.
- LBCL-IRF4 cases are primarily germinal center–derived B-cell lymphomas. They commonly present with nodal involvement of the head and neck (particularly the Waldeyer ring) and less commonly in the gastrointestinal tract.
- LBCL-IRF4 shows strong IRF4 expression. In a study of 17 cases, the most frequently altered genes were CARD11 (35%) and CCND3 (24%).
- LBCL-IRF4 appears to be a low stage at diagnosis and is associated with a favorable prognosis compared with diffuse large B-cell lymphoma cases lacking this abnormality.
High-grade B-cell lymphoma, NOS, is defined as a clinically aggressive B-cell lymphoma that lacks MYC plus BCL2 and/or BCL6 rearrangements. In addition, this entity does not meet criteria for diffuse large B-cell lymphoma, NOS, or Burkitt lymphoma.
- High-grade B-cell lymphoma, NOS, is a biologically heterogeneous disease. In a study of eight cases of pediatric high-grade B-cell lymphoma, NOS, four had variant profiles similar to that of Burkitt lymphoma (e.g., MYC rearrangements and variants in CCND3, ID3, and DDX3X). The remaining cases lacked MYC rearrangements and had variant profiles closer to GCB-DLBCL (e.g., TNFRSF14, CARD11 and EZH2 variants), and lacked MYC translocations.
For information about the treatment of childhood diffuse large B-cell lymphoma, see Childhood Non-Hodgkin Lymphoma Treatment.
Primary mediastinal B-cell lymphoma
Genomics of primary mediastinal B-cell lymphoma
Primary mediastinal B-cell lymphoma was previously considered a subtype of diffuse large B-cell lymphoma, but is now a separate entity in the World Health Organization (WHO) classification. These tumors arise in the mediastinum from thymic B cells and show a diffuse large cell proliferation with sclerosis that compartmentalizes neoplastic cells.
Primary mediastinal B-cell lymphoma can be very difficult to distinguish morphologically from the following types of lymphoma:
- Diffuse large B-cell lymphoma: Cell surface markers in primary mediastinal B-cell lymphoma are similar to the ones seen in diffuse large B-cell lymphoma (i.e., CD19, CD20, CD22, CD79a, and PAX-5). However, primary mediastinal B-cell lymphoma may display cytoplasmic immunoglobulins, and CD30 expression is commonly present.
- Hodgkin lymphoma: Primary mediastinal B-cell lymphoma may be difficult to distinguish from Hodgkin lymphoma clinically and morphologically, especially with small mediastinal biopsies because of extensive sclerosis and necrosis.
Primary mediastinal B-cell lymphoma has distinctive gene expression and variant profiles compared with diffuse large B-cell lymphoma. However, its gene expression and variant profiles have features similar to those seen in Hodgkin lymphoma. Primary mediastinal B-cell lymphoma is also associated with a distinctive constellation of chromosomal aberrations compared with other NHL subtypes. Because primary mediastinal B-cell lymphoma is primarily a cancer of adolescents and young adults, the genomic findings are presented without regard to age.
- Multiple genomic alterations contribute to immune evasion in primary mediastinal B-cell lymphoma:
- Structural rearrangements and copy number gains at chromosome 9p24 are common in primary mediastinal B-cell lymphoma. This region encodes the immune checkpoint genes CD274 (PDL1) and PDCD1LG2. The genomic alterations lead to increased expression of these checkpoint proteins.
- Genomic alterations in CIITA, which is the master transcriptional regulator of major histocompatibility complex (MHC) class II expression, are common in primary mediastinal B-cell lymphoma. These alterations lead to loss of MHC class II expression.
- Approximately 50% of primary mediastinal B-cell lymphoma cases show variants or focal copy number losses in B2M, the gene that encodes beta-2-microglobulin (the invariant chain of the MHC class I). These alterations lead to reduced expression of MHC class I.
- Genomic alterations involving genes of the JAK-STAT pathway are observed in most cases of primary mediastinal B-cell lymphoma.
- STAT6 is altered in approximately 40% of primary mediastinal B-cell lymphoma cases.
- The chromosome 9p region that shows gains and amplification in primary mediastinal B-cell lymphoma encodes JAK2, which activates the STAT pathway.
- SOCS1, a negative regulator of JAK-STAT signaling, is inactivated in approximately 50% to 60% of primary mediastinal B-cell lymphoma cases by either variant or gene deletion.
- The IL4R gene shows activating variants in approximately 20% to 30% of primary mediastinal B-cell lymphoma cases. IL4R activation leads to increased JAK-STAT pathway activity.
- Genomic alterations leading to NF-ĸB activation are also common in primary mediastinal B-cell lymphoma. These include copy number gains and amplifications at 2p16.1, a region that encodes BCL11A and REL. Genes encoding negative regulators of NF-kB signaling (e.g., TNFAIP3 and NFKBIE) show inactivating variants in primary mediastinal B-cell lymphoma.
- Other genes that are altered in primary mediastinal B-cell lymphoma include ZNF217, XPO1, and EZH2.
For information about the treatment of childhood primary mediastinal B-cell lymphoma, see Childhood Non-Hodgkin Lymphoma Treatment.
Lymphoblastic Lymphoma
Genomics of lymphoblastic lymphoma
Lymphoblastic lymphomas are usually positive for terminal deoxynucleotidyl transferase. More than 75% of cases have a T-cell immunophenotype and the remaining cases have a precursor B-cell phenotype.
As opposed to pediatric T-cell acute lymphoblastic leukemia (T-ALL), the molecular biology and chromosomal abnormalities of pediatric lymphoblastic lymphoma are not as well characterized. Many genomic alterations that occur in T-ALL also occur in T-cell lymphoblastic lymphoma. Examples include the following:
- NOTCH1 and FBXW7 variants (which also induce NOTCH pathway signaling) are common in T-ALL. NOTCH1 and FBXW7 variants are also observed in approximately 60% to 65% and 15% to 25% of T-cell lymphoblastic lymphoma cases, respectively.
- CDKN2A at chromosome 9p21 is commonly altered in both T-ALL and in T-cell lymphoblastic lymphoma, with approximately three-fourths of each showing deletions of this gene locus.
- Loss of heterozygosity at chromosome 6q is observed in approximately 15% of T-ALL cases.
- PTEN variants are observed in approximately 15% of T-ALL cases and in a comparable percentage of T-lymphoblastic lymphoma cases.
- KMT2D variants are observed in approximately 10% of T-lymphoblastic lymphoma cases. Other genes associated with epigenetics that are altered in T-ALL include PHF6 and KMT2C.
For the genomic alterations described above, NOTCH1 and FBXW7 variants may confer a more favorable prognosis for patients with T-cell lymphoblastic lymphoma. In contrast, loss of heterozygosity at chromosome 6q, PTEN variants, and KMT2D variants may be associated with an inferior prognosis. For example, one study noted that the presence of a KMT2D and/or PTEN variant was associated with a high risk of relapse in patients with wild-type NOTCH1 or FBXW7, but these variants were not associated with an increased risk of relapse in patients with variants in NOTCH1 or FBXW7. Studies with larger numbers of patients are needed to better define the critical genomic determinants of outcome for patients with T-cell lymphoblastic lymphoma.
There have been few studies of the genomic characteristics of B-lymphoblastic lymphoma. One report described copy number alterations for pediatric B-lymphoblastic lymphoma cases. The study noted that some gene deletions that are common in B-ALL (e.g., CDKN2A, IKZF1, and PAX5) appeared to occur with appreciable frequency in B-lymphoblastic lymphoma.
For information about the treatment of childhood lymphoblastic lymphoma, see Childhood Non-Hodgkin Lymphoma Treatment.
Anaplastic Large Cell Lymphoma
Genomics of anaplastic large cell lymphoma
While mature T cell is the predominant immunophenotype of anaplastic large cell lymphoma, null-cell disease (i.e., no T-cell, B-cell, or natural killer-cell surface antigen expression) does occur. The World Health Organization (WHO) classifies anaplastic large cell lymphoma as a subtype of peripheral T-cell lymphoma.
All anaplastic large cell lymphoma cases are CD30-positive. More than 90% of pediatric anaplastic large cell lymphoma cases have a chromosomal rearrangement involving the ALK gene. About 85% of these chromosomal rearrangements will be t(2;5)(p23;q35), leading to the expression of the NPM::ALK fusion protein. The other 15% of cases are composed of variant ALK translocations. The anti-ALK immunohistochemical staining pattern is quite specific for the type of ALK translocation. Cytoplasm and nuclear ALK staining is associated with NPM::ALK fusion proteins, whereas cytoplasmic staining of ALK is only associated with the variant ALK translocations, as shown in Table 4.
| Gene Fusion | Partner Chromosome Location | Frequency of Gene Fusion |
|---|---|---|
| aAdapted from Tsuyama et al. | ||
| NPM::ALK | 5q36.1 | Approximately 80% |
| TPM3::ALK | 1p23 | Approximately 15% |
| ALO17::ALK | 17q25.3 | Rare |
| ATIC::ALK | 2q35 | Rare |
| CLTC::ALK | 17q23 | Rare |
| MSN::ALK | Xp11.1 | Rare |
| MYH9::ALK | 22q13.1 | Rare |
| TFG::ALK | 3q12.2 | Rare |
| TPM4::ALK | 19p13 | Rare |
| TRAF1::ALK | 9q33.2 | Rare |
In adults, ALK-positive anaplastic large cell lymphoma is viewed differently from other peripheral T-cell lymphomas because prognosis tends to be superior. Also, adult patients with ALK-negative anaplastic large cell lymphoma have an inferior outcome compared with patients who have ALK-positive disease. In children, however, this difference in outcome between ALK-positive and ALK-negative disease has not been demonstrated. In addition, no correlation has been found between outcome and the specific ALK-translocation type.
One European series included 375 children and adolescents with systemic ALK-positive anaplastic large cell lymphoma. The presence of a small cell or lymphohistiocytic component was observed in 32% of patients, and it was significantly associated with a high risk of failure in the multivariate analysis, controlling for clinical characteristics (hazard ratio, 2.0; P = .002). The prognostic implication of the small cell variant of anaplastic large cell lymphoma was also shown in the COG-ANHL0131 (NCT00059839) study, despite using a different chemotherapy backbone.
For information about the treatment of childhood anaplastic large cell lymphoma, see Childhood Non-Hodgkin Lymphoma Treatment.
Pediatric-Type Follicular Lymphoma
Genomics of pediatric-type follicular lymphoma
Pediatric-type follicular lymphoma and nodal marginal zone lymphoma are rare indolent B-cell lymphomas that are clinically and molecularly distinct from these tumor types in adults.
- The pediatric types lack BCL2 and IRF4 rearrangements, resulting in IRF4 expression.
- BCL6 and MYC rearrangements are also not present in pediatric-type follicular lymphoma.
- TNFSFR14 variants are common in pediatric-type follicular lymphoma. These variants appear to occur with similar frequency in adult follicular lymphoma.
- MAP2K1 variants, which are uncommon in adults, are observed in as many as 43% of pediatric-type follicular lymphoma cases. Other genes (e.g., MAPK1 and RRAS) have been found to be altered in cases without MAP2K1 variants. This finding suggests that the MAP kinase pathway is important in the pathogenesis of pediatric-type follicular lymphoma.
- IRF8 variants, KMT2C variants, and abnormalities in chromosome 1p have also been observed in pediatric-type follicular lymphoma.
For information about the treatment of pediatric-type follicular lymphoma, see Childhood Non-Hodgkin Lymphoma Treatment.
References
- Leoncini L, Raphael M, Stein H, et al.: Burkitt lymphoma. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th rev. ed. International Agency for Research on Cancer, 2017, pp 330-4.
- Perkins SL, Lones MA, Davenport V, et al.: B-Cell non-Hodgkin's lymphoma in children and adolescents: surface antigen expression and clinical implications for future targeted bioimmune therapy: a children's cancer group report. Clin Adv Hematol Oncol 1 (5): 314-7, 2003.
- Miles RR, Cairo MS, Satwani P, et al.: Immunophenotypic identification of possible therapeutic targets in paediatric non-Hodgkin lymphomas: a children's oncology group report. Br J Haematol 138 (4): 506-12, 2007.
- Gualco G, Weiss LM, Harrington WJ, et al.: Nodal diffuse large B-cell lymphomas in children and adolescents: immunohistochemical expression patterns and c-MYC translocation in relation to clinical outcome. Am J Surg Pathol 33 (12): 1815-22, 2009.
- Grande BM, Gerhard DS, Jiang A, et al.: Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood 133 (12): 1313-1324, 2019.
- López C, Kleinheinz K, Aukema SM, et al.: Genomic and transcriptomic changes complement each other in the pathogenesis of sporadic Burkitt lymphoma. Nat Commun 10 (1): 1459, 2019.
- Schmitz R, Young RM, Ceribelli M, et al.: Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 490 (7418): 116-20, 2012.
- Richter J, Schlesner M, Hoffmann S, et al.: Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat Genet 44 (12): 1316-20, 2012.
- Havelange V, Pepermans X, Ameye G, et al.: Genetic differences between paediatric and adult Burkitt lymphomas. Br J Haematol 173 (1): 137-44, 2016.
- Rohde M, Bonn BR, Zimmermann M, et al.: Relevance of ID3-TCF3-CCND3 pathway mutations in pediatric aggressive B-cell lymphoma treated according to the non-Hodgkin Lymphoma Berlin-Frankfurt-Münster protocols. Haematologica 102 (6): 1091-1098, 2017.
- Chakraborty AA, Scuoppo C, Dey S, et al.: A common functional consequence of tumor-derived mutations within c-MYC. Oncogene 34 (18): 2406-9, 2015.
- Masqué-Soler N, Szczepanowski M, Kohler CW, et al.: Clinical and pathological features of Burkitt lymphoma showing expression of BCL2--an analysis including gene expression in formalin-fixed paraffin-embedded tissue. Br J Haematol 171 (4): 501-8, 2015.
- Kluin PM, Harris NL, Stein H: B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th rev. ed. International Agency for Research on Cancer, 2017, pp 314-6.
- Alaggio R, Amador C, Anagnostopoulos I, et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 36 (7): 1720-1748, 2022.
- Wagener R, Seufert J, Raimondi F, et al.: The mutational landscape of Burkitt-like lymphoma with 11q aberration is distinct from that of Burkitt lymphoma. Blood 133 (9): 962-966, 2019.
- Gonzalez-Farre B, Ramis-Zaldivar JE, Salmeron-Villalobos J, et al.: Burkitt-like lymphoma with 11q aberration: a germinal center-derived lymphoma genetically unrelated to Burkitt lymphoma. Haematologica 104 (9): 1822-1829, 2019.
- Au-Yeung RKH, Arias Padilla L, Zimmermann M, et al.: Experience with provisional WHO-entities large B-cell lymphoma with IRF4-rearrangement and Burkitt-like lymphoma with 11q aberration in paediatric patients of the NHL-BFM group. Br J Haematol 190 (5): 753-763, 2020.
- Chapuy B, Stewart C, Dunford AJ, et al.: Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 24 (5): 679-690, 2018.
- Schmitz R, Wright GW, Huang DW, et al.: Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med 378 (15): 1396-1407, 2018.
- Oschlies I, Klapper W, Zimmermann M, et al.: Diffuse large B-cell lymphoma in pediatric patients belongs predominantly to the germinal-center type B-cell lymphomas: a clinicopathologic analysis of cases included in the German BFM (Berlin-Frankfurt-Munster) Multicenter Trial. Blood 107 (10): 4047-52, 2006.
- Miles RR, Raphael M, McCarthy K, et al.: Pediatric diffuse large B-cell lymphoma demonstrates a high proliferation index, frequent c-Myc protein expression, and a high incidence of germinal center subtype: Report of the French-American-British (FAB) international study group. Pediatr Blood Cancer 51 (3): 369-74, 2008.
- Ramis-Zaldivar JE, Gonzalez-Farré B, Balagué O, et al.: Distinct molecular profile of IRF4-rearranged large B-cell lymphoma. Blood 135 (4): 274-286, 2020.
- Klapper W, Kreuz M, Kohler CW, et al.: Patient age at diagnosis is associated with the molecular characteristics of diffuse large B-cell lymphoma. Blood 119 (8): 1882-7, 2012.
- Klapper W, Szczepanowski M, Burkhardt B, et al.: Molecular profiling of pediatric mature B-cell lymphoma treated in population-based prospective clinical trials. Blood 112 (4): 1374-81, 2008.
- Deffenbacher KE, Iqbal J, Sanger W, et al.: Molecular distinctions between pediatric and adult mature B-cell non-Hodgkin lymphomas identified through genomic profiling. Blood 119 (16): 3757-66, 2012.
- Poirel HA, Cairo MS, Heerema NA, et al.: Specific cytogenetic abnormalities are associated with a significantly inferior outcome in children and adolescents with mature B-cell non-Hodgkin's lymphoma: results of the FAB/LMB 96 international study. Leukemia 23 (2): 323-31, 2009.
- Pittaluga S, Harris NL, Siebert R, et al.: Large B-cell lymphoma with IRF4 rearrangement. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th rev. ed. International Agency for Research on Cancer, 2017, pp 280-1.
- Chisholm KM, Mohlman J, Liew M, et al.: IRF4 translocation status in pediatric follicular and diffuse large B-cell lymphoma patients enrolled in Children's Oncology Group trials. Pediatr Blood Cancer 66 (8): e27770, 2019.
- Salaverria I, Philipp C, Oschlies I, et al.: Translocations activating IRF4 identify a subtype of germinal center-derived B-cell lymphoma affecting predominantly children and young adults. Blood 118 (1): 139-47, 2011.
- Liu Q, Salaverria I, Pittaluga S, et al.: Follicular lymphomas in children and young adults: a comparison of the pediatric variant with usual follicular lymphoma. Am J Surg Pathol 37 (3): 333-43, 2013.
- Jiang XN, Yu F, Xue T, et al.: IRF4 rearrangement may predict favorable prognosis in children and young adults with primary head and neck large B-cell lymphoma. Cancer Med 12 (9): 10684-10693, 2023.
- Kluin PM, Harris NL, Stein H, et al.: High-grade B-cell lymphoma. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th rev. ed. International Agency for Research on Cancer, 2017, pp 335-41.
- Jaffe ES, Harris NL, Stein H, et al.: Introduction and overview of the classification of the lymphoid neoplasms. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 157-66.
- Rosenwald A, Wright G, Leroy K, et al.: Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 198 (6): 851-62, 2003.
- Savage KJ, Monti S, Kutok JL, et al.: The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood 102 (12): 3871-9, 2003.
- Mottok A, Hung SS, Chavez EA, et al.: Integrative genomic analysis identifies key pathogenic mechanisms in primary mediastinal large B-cell lymphoma. Blood 134 (10): 802-813, 2019.
- Green MR, Monti S, Rodig SJ, et al.: Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 116 (17): 3268-77, 2010.
- Twa DD, Chan FC, Ben-Neriah S, et al.: Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood 123 (13): 2062-5, 2014.
- Chong LC, Twa DD, Mottok A, et al.: Comprehensive characterization of programmed death ligand structural rearrangements in B-cell non-Hodgkin lymphomas. Blood 128 (9): 1206-13, 2016.
- Chapuy B, Stewart C, Dunford AJ, et al.: Genomic analyses of PMBL reveal new drivers and mechanisms of sensitivity to PD-1 blockade. Blood 134 (26): 2369-2382, 2019.
- Mottok A, Woolcock B, Chan FC, et al.: Genomic Alterations in CIITA Are Frequent in Primary Mediastinal Large B Cell Lymphoma and Are Associated with Diminished MHC Class II Expression. Cell Rep 13 (7): 1418-1431, 2015.
- Viganò E, Gunawardana J, Mottok A, et al.: Somatic IL4R mutations in primary mediastinal large B-cell lymphoma lead to constitutive JAK-STAT signaling activation. Blood 131 (18): 2036-2046, 2018.
- Bea S, Zettl A, Wright G, et al.: Diffuse large B-cell lymphoma subgroups have distinct genetic profiles that influence tumor biology and improve gene-expression-based survival prediction. Blood 106 (9): 3183-90, 2005.
- Oschlies I, Burkhardt B, Salaverria I, et al.: Clinical, pathological and genetic features of primary mediastinal large B-cell lymphomas and mediastinal gray zone lymphomas in children. Haematologica 96 (2): 262-8, 2011.
- Melzner I, Bucur AJ, Brüderlein S, et al.: Biallelic mutation of SOCS-1 impairs JAK2 degradation and sustains phospho-JAK2 action in the MedB-1 mediastinal lymphoma line. Blood 105 (6): 2535-42, 2005.
- Mestre C, Rubio-Moscardo F, Rosenwald A, et al.: Homozygous deletion of SOCS1 in primary mediastinal B-cell lymphoma detected by CGH to BAC microarrays. Leukemia 19 (6): 1082-4, 2005.
- Neth O, Seidemann K, Jansen P, et al.: Precursor B-cell lymphoblastic lymphoma in childhood and adolescence: clinical features, treatment, and results in trials NHL-BFM 86 and 90. Med Pediatr Oncol 35 (1): 20-7, 2000.
- Liu Y, Easton J, Shao Y, et al.: The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet 49 (8): 1211-1218, 2017.
- Bonn BR, Rohde M, Zimmermann M, et al.: Incidence and prognostic relevance of genetic variations in T-cell lymphoblastic lymphoma in childhood and adolescence. Blood 121 (16): 3153-60, 2013.
- Burkhardt B, Moericke A, Klapper W, et al.: Pediatric precursor T lymphoblastic leukemia and lymphoblastic lymphoma: Differences in the common regions with loss of heterozygosity at chromosome 6q and their prognostic impact. Leuk Lymphoma 49 (3): 451-61, 2008.
- Balbach ST, Makarova O, Bonn BR, et al.: Proposal of a genetic classifier for risk group stratification in pediatric T-cell lymphoblastic lymphoma reveals differences from adult T-cell lymphoblastic leukemia. Leukemia 30 (4): 970-3, 2016.
- Khanam T, Sandmann S, Seggewiss J, et al.: Integrative genomic analysis of pediatric T-cell lymphoblastic lymphoma reveals candidates of clinical significance. Blood 137 (17): 2347-2359, 2021.
- Callens C, Baleydier F, Lengline E, et al.: Clinical impact of NOTCH1 and/or FBXW7 mutations, FLASH deletion, and TCR status in pediatric T-cell lymphoblastic lymphoma. J Clin Oncol 30 (16): 1966-73, 2012.
- Meyer JA, Zhou D, Mason CC, et al.: Genomic characterization of pediatric B-lymphoblastic lymphoma and B-lymphoblastic leukemia using formalin-fixed tissues. Pediatr Blood Cancer 64 (7): , 2017.
- Swerdlow SH, Campo E, Pileri SA, et al.: The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127 (20): 2375-90, 2016.
- Duyster J, Bai RY, Morris SW: Translocations involving anaplastic lymphoma kinase (ALK). Oncogene 20 (40): 5623-37, 2001.
- Tsuyama N, Sakamoto K, Sakata S, et al.: Anaplastic large cell lymphoma: pathology, genetics, and clinical aspects. J Clin Exp Hematop 57 (3): 120-142, 2017.
- Savage KJ, Harris NL, Vose JM, et al.: ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood 111 (12): 5496-504, 2008.
- Vose J, Armitage J, Weisenburger D, et al.: International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol 26 (25): 4124-30, 2008.
- Stein H, Foss HD, Dürkop H, et al.: CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood 96 (12): 3681-95, 2000.
- Lamant L, McCarthy K, d'Amore E, et al.: Prognostic impact of morphologic and phenotypic features of childhood ALK-positive anaplastic large-cell lymphoma: results of the ALCL99 study. J Clin Oncol 29 (35): 4669-76, 2011.
- Alexander S, Kraveka JM, Weitzman S, et al.: Advanced stage anaplastic large cell lymphoma in children and adolescents: results of ANHL0131, a randomized phase III trial of APO versus a modified regimen with vinblastine: a report from the children's oncology group. Pediatr Blood Cancer 61 (12): 2236-42, 2014.
- Jaffe ES, Harris NL, Siebert R: Paediatric-type follicular lymphoma. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th rev. ed. International Agency for Research on Cancer, 2017, pp 278-9.
- Launay E, Pangault C, Bertrand P, et al.: High rate of TNFRSF14 gene alterations related to 1p36 region in de novo follicular lymphoma and impact on prognosis. Leukemia 26 (3): 559-62, 2012.
- Schmidt J, Gong S, Marafioti T, et al.: Genome-wide analysis of pediatric-type follicular lymphoma reveals low genetic complexity and recurrent alterations of TNFRSF14 gene. Blood 128 (8): 1101-11, 2016.
- Louissaint A, Schafernak KT, Geyer JT, et al.: Pediatric-type nodal follicular lymphoma: a biologically distinct lymphoma with frequent MAPK pathway mutations. Blood 128 (8): 1093-100, 2016.
- Schmidt J, Ramis-Zaldivar JE, Nadeu F, et al.: Mutations of MAP2K1 are frequent in pediatric-type follicular lymphoma and result in ERK pathway activation. Blood 130 (3): 323-327, 2017.
- Ozawa MG, Bhaduri A, Chisholm KM, et al.: A study of the mutational landscape of pediatric-type follicular lymphoma and pediatric nodal marginal zone lymphoma. Mod Pathol 29 (10): 1212-20, 2016.
- Lim S, Lim KY, Koh J, et al.: Pediatric-Type Indolent B-Cell Lymphomas With Overlapping Clinical, Pathologic, and Genetic Features. Am J Surg Pathol 46 (10): 1397-1406, 2022.
Hodgkin Lymphoma
Genomics of Classical Hodgkin Lymphoma
Classical Hodgkin lymphoma has a gene expression and variant profile that differs from that of other lymphomas. The exception is primary mediastinal B-cell lymphoma, which shares many genomic and cytogenetic characteristics with Hodgkin lymphoma. Characterization of genomic alterations for Hodgkin lymphoma is challenging because malignant Hodgkin and Reed-Sternberg (HRS) cells make up only a small percentage of the overall tumor mass. Because of this finding, special methods, such as microdissection of HRS cells or flow cytometry cell sorting, are required before applying molecular analysis methods. Hodgkin lymphoma genomic alterations can also be assessed using special sequencing methods applied to circulating cell-free DNA (cfDNA) in peripheral blood of patients with Hodgkin lymphoma.
The genomic alterations observed in Hodgkin lymphoma fall into several categories, including immune evasion alterations, JAK-STAT pathway alterations, alterations leading to nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kappaB) activation, and others:
- Multiple genomic alterations contribute to immune evasion in Hodgkin lymphoma.
- Copy number gain or amplification at chromosome 9p24 is observed in most cases of Hodgkin lymphoma. This region encodes the immune checkpoint genes CD274 (encoding PD-L1) and PDCD1LG2 (encoding PD-L2). These genomic alterations lead to increased expression of these checkpoint proteins.
- Gene fusions involving CIITA, which is the master transcriptional regulator of major histocompatibility complex (MHC) class II expression, were reported in 15% of Hodgkin lymphoma cases. Similar alterations are found in primary mediastinal B-cell lymphoma, and they lead to decreased CIITA protein expression and loss of MHC class II expression.
- Beta-2-microglobulin (the invariant chain of the MHC class I) frequently shows decreased/absent expression in HRS cells, with accompanying decreased MHC class I expression. Inactivating variants in B2M, the gene that encodes beta-2-microglobulin, are common in Hodgkin lymphoma and lead to reduced expression of MHC class I. Inactivating variants in B2M occur more frequently in Epstein-Barr virus (EBV)-negative Hodgkin lymphoma than in EBV-positive Hodgkin lymphoma, which explains the higher rates of beta-2 microglobulin and MHC class I expression for EBV-positive Hodgkin lymphoma, compared with EBV-negative Hodgkin lymphoma.
- Genomic alterations involving genes in the JAK-STAT pathway are observed in most cases of Hodgkin lymphoma. Genes in the JAK-STAT pathway for which genomic alterations are reported include:
- SOCS1, a negative regulator of JAK-STAT signaling, is inactivated by variants in 60% to 70% of Hodgkin lymphoma cases. In a study of pediatric Hodgkin lymphoma using cfDNA collected before treatment, SOCS1 was the most frequently altered gene, with variants in 60% of all cases and approximately 80% of cases in which genomic alterations were detected in cfDNA.
- Activating STAT6 variants occurring at hot spots in the DNA-binding domain are observed in approximately 30% of Hodgkin lymphoma cases.
- The chromosome 9p region that contains CD274 and PDCD1LG2, which shows gains and amplifications in Hodgkin lymphoma, also contains JAK2. Chromosome 9p gain/amplification is thought to further augment JAK-STAT pathway signaling.
- Inactivating variants in PTPN1, a phosphatase that inhibits JAK-STAT pathway signaling, were observed in approximately 20% of Hodgkin lymphoma cases.
- Variants in other genes affecting JAK-STAT pathway signaling have also been reported, including JAK1, STAT3, STAT5B, and CSF2RB.
- Genomic alterations leading to NF-kappaB activation are also common in Hodgkin lymphoma.
- The REL gene at chromosome 2p16.1 shows genomic gain or amplification in approximately one-third of Hodgkin lymphoma cases.
- EBV-positive Hodgkin lymphoma expresses the EBV latent membrane protein 1 (LMP1) at the cell surface. This protein acts like a constitutively activated receptor of the TNF receptor family to cause activation of the NF-kappaB pathway.
- Inactivating variants in genes that inhibit NF-kappaB pathway signaling, including TNFAIP3, NFKBIA, and NFKBIE, are common in Hodgkin lymphoma. Inactivation of the gene products for these genes leads to NF-kappaB pathway activation. TNFAIP3 is the most commonly altered inhibitor of NF-kappaB pathway signaling, and loss of function alterations occur by either variants or by focal 6q23.3 or arm-level 6q loss. TNFAIP3 genomic alterations are much more common in EBV-negative Hodgkin lymphoma than in EBV-positive Hodgkin lymphoma, suggesting that LMP1 expression in EBV-positive Hodgkin lymphoma obviates the need for TNFAIP3 loss of function.
- Other genes with variants in Hodgkin lymphoma include XPO1, RBM38, ACTB, ARID1A, and GNA13.
- Hodgkin lymphoma is derived from a B-cell progenitor, and HRS cells generally do not express B-cell surface antigens. HRS cells do have immunoglobulin (Ig) heavy and light chain V gene rearrangements typical of B cells. Although Ig genes have undergone rearrangements in HRS cells, the rearrangements are nonproductive and B-cell receptor is not expressed.
Genomics of Nodular Lymphocyte-Predominant Hodgkin Lymphoma
The lymphocyte-predominant (LP) cells of nodular lymphocyte-predominant Hodgkin lymphoma have distinctive genomic characteristics compared with the HRS cells of Hodgkin lymphoma. As with Hodgkin lymphoma, genomic characterization is complicated by the low percentage of malignant cells within a tumor mass.
- LP cells express B-cell antigens (e.g., CD19, CD20, CD22, and CD79A) and B-cell transcription factors (e.g., OCT2 and BOB1).
- The expression of Bcl-6 and the presence of somatic hypervariants in the variable region of rearranged Ig heavy chain genes point to a germinal center derivation for LP cells.
- IgD expression connotes a distinct type of nodular lymphocyte-predominant Hodgkin lymphoma that is associated with a very high male-to-female ratio (>10:1). An evaluation of the antigenic specificity of the B-cell receptor in cases of IgD-positive nodular lymphocyte-predominant Hodgkin lymphoma found that in 7 of 8 cases (6 of 8 patients aged ≤18 years) the B-cell receptor recognized the DNA-directed RNA polymerase (RpoC) from Moraxella catarrhalis. High-titer, light-chain-restricted anti-RpoC IgG1 serum-antibodies were observed in these patients. In addition, MID/hag is a superantigen expressed by M. catarrhalis that binds to the Fc domain of IgD and activates IgD-positive B cells. These observations support a role for M. catarrhalis in the development and maintenance of IgD-positive nodular lymphocyte-predominant Hodgkin lymphoma.
- Genomic analysis of nodular lymphocyte-predominant Hodgkin lymphoma is limited to a small number of patients using gene panels to evaluate microdissected specimens containing LP cells. Genes with recurring variants include SOCS1 (an inhibitor of JAK-STAT pathway signaling), DUSP2 (a dual specificity phosphatase that is a negative regulator of the MAP kinase pathway), JUNB (a transcription factor in the activator protein-1 family), and SGK1 (a serine-threonine kinase).
For information about the treatment of childhood Hodgkin lymphoma, see Childhood Hodgkin Lymphoma Treatment.
References
- Mottok A, Hung SS, Chavez EA, et al.: Integrative genomic analysis identifies key pathogenic mechanisms in primary mediastinal large B-cell lymphoma. Blood 134 (10): 802-813, 2019.
- Wienand K, Chapuy B, Stewart C, et al.: Genomic analyses of flow-sorted Hodgkin Reed-Sternberg cells reveal complementary mechanisms of immune evasion. Blood Adv 3 (23): 4065-4080, 2019.
- Tiacci E, Ladewig E, Schiavoni G, et al.: Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood 131 (22): 2454-2465, 2018.
- Spina V, Bruscaggin A, Cuccaro A, et al.: Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood 131 (22): 2413-2425, 2018.
- Roemer MG, Advani RH, Ligon AH, et al.: PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J Clin Oncol 34 (23): 2690-7, 2016.
- Roemer MGM, Redd RA, Cader FZ, et al.: Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Programmed Death 1 Blockade in Classic Hodgkin Lymphoma. J Clin Oncol 36 (10): 942-950, 2018.
- Steidl C, Shah SP, Woolcock BW, et al.: MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 471 (7338): 377-81, 2011.
- Mottok A, Woolcock B, Chan FC, et al.: Genomic Alterations in CIITA Are Frequent in Primary Mediastinal Large B Cell Lymphoma and Are Associated with Diminished MHC Class II Expression. Cell Rep 13 (7): 1418-1431, 2015.
- Roemer MG, Advani RH, Redd RA, et al.: Classical Hodgkin Lymphoma with Reduced β2M/MHC Class I Expression Is Associated with Inferior Outcome Independent of 9p24.1 Status. Cancer Immunol Res 4 (11): 910-916, 2016.
- Reichel J, Chadburn A, Rubinstein PG, et al.: Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood 125 (7): 1061-72, 2015.
- Desch AK, Hartung K, Botzen A, et al.: Genotyping circulating tumor DNA of pediatric Hodgkin lymphoma. Leukemia 34 (1): 151-166, 2020.
- Green MR, Monti S, Rodig SJ, et al.: Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 116 (17): 3268-77, 2010.
- Gunawardana J, Chan FC, Telenius A, et al.: Recurrent somatic mutations of PTPN1 in primary mediastinal B cell lymphoma and Hodgkin lymphoma. Nat Genet 46 (4): 329-35, 2014.
- Steidl C, Telenius A, Shah SP, et al.: Genome-wide copy number analysis of Hodgkin Reed-Sternberg cells identifies recurrent imbalances with correlations to treatment outcome. Blood 116 (3): 418-27, 2010.
- Gires O, Zimber-Strobl U, Gonnella R, et al.: Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J 16 (20): 6131-40, 1997.
- Schmitz R, Hansmann ML, Bohle V, et al.: TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med 206 (5): 981-9, 2009.
- Küppers R, Rajewsky K, Zhao M, et al.: Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A 91 (23): 10962-6, 1994.
- Kanzler H, Küppers R, Helmes S, et al.: Hodgkin and Reed-Sternberg-like cells in B-cell chronic lymphocytic leukemia represent the outgrowth of single germinal-center B-cell-derived clones: potential precursors of Hodgkin and Reed-Sternberg cells in Hodgkin's disease. Blood 95 (3): 1023-31, 2000.
- Shankar A, Daw S: Nodular lymphocyte predominant Hodgkin lymphoma in children and adolescents--a comprehensive review of biology, clinical course and treatment options. Br J Haematol 159 (3): 288-98, 2012.
- Stein H, Marafioti T, Foss HD, et al.: Down-regulation of BOB.1/OBF.1 and Oct2 in classical Hodgkin disease but not in lymphocyte predominant Hodgkin disease correlates with immunoglobulin transcription. Blood 97 (2): 496-501, 2001.
- Braeuninger A, Küppers R, Strickler JG, et al.: Hodgkin and Reed-Sternberg cells in lymphocyte predominant Hodgkin disease represent clonal populations of germinal center-derived tumor B cells. Proc Natl Acad Sci U S A 94 (17): 9337-42, 1997.
- Falini B, Bigerna B, Pasqualucci L, et al.: Distinctive expression pattern of the BCL-6 protein in nodular lymphocyte predominance Hodgkin's disease. Blood 87 (2): 465-71, 1996.
- Huppmann AR, Nicolae A, Slack GW, et al.: EBV may be expressed in the LP cells of nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) in both children and adults. Am J Surg Pathol 38 (3): 316-24, 2014.
- Prakash S, Fountaine T, Raffeld M, et al.: IgD positive L&H cells identify a unique subset of nodular lymphocyte predominant Hodgkin lymphoma. Am J Surg Pathol 30 (5): 585-92, 2006.
- Thurner L, Hartmann S, Neumann F, et al.: Role of Specific B-Cell Receptor Antigens in Lymphomagenesis. Front Oncol 10: 604685, 2020.
- Hartmann S, Schuhmacher B, Rausch T, et al.: Highly recurrent mutations of SGK1, DUSP2 and JUNB in nodular lymphocyte predominant Hodgkin lymphoma. Leukemia 30 (4): 844-53, 2016.
- Mottok A, Renné C, Willenbrock K, et al.: Somatic hypermutation of SOCS1 in lymphocyte-predominant Hodgkin lymphoma is accompanied by high JAK2 expression and activation of STAT6. Blood 110 (9): 3387-90, 2007.
- Schuhmacher B, Bein J, Rausch T, et al.: JUNB, DUSP2, SGK1, SOCS1 and CREBBP are frequently mutated in T-cell/histiocyte-rich large B-cell lymphoma. Haematologica 104 (2): 330-337, 2019.
Central Nervous System Tumors
Central nervous system (CNS) tumors include gliomas (including astrocytomas), glioneuronal tumors, neuronal tumors, CNS atypical teratoid/rhabdoid tumors, medulloblastomas, nonmedulloblastoma embryonal tumors, pineal tumors, and ependymomas.
The terminology of the 2021 World Health Organization (WHO) Classification of Tumors of the Central Nervous System is used below. The 2021 WHO CNS classification advances the role of molecular diagnostics in CNS tumor classification, and it includes multiple major changes from the previous 2016 WHO classification.
Astrocytomas, Other Gliomas, and Glioneuronal/Neuronal Tumors
This category includes, among other diagnoses, pediatric-type diffuse low-grade gliomas, pediatric-type diffuse high-grade gliomas, circumscribed astrocytic gliomas, glioneuronal tumors, and neuronal tumors.
For pediatric-type diffuse gliomas, rearrangements in the MYB family of transcription factors (MYB and MYBL1) are the most commonly reported genomic alteration in low-grade tumors. Other alterations observed include FGFR1 alterations (primarily duplications involving the tyrosine kinase domain), BRAF alterations, NF1 variants, and RAS family variants. IDH1 variants, which are the most common genomic alteration in adult-type diffuse astrocytomas, are uncommon in children with diffuse astrocytomas and, when present, are observed almost exclusively in older adolescents.
The diffuse midline glioma, H3 K27M-altered, category includes tumors previously classified as diffuse intrinsic pontine glioma (DIPG). Most of the data is derived from experience with DIPG. This category also includes gliomas with the H3 K27M variant arising in midline structures such as the thalamus.
Selected cancer susceptibility syndromes associated with pediatric glioma
Neurofibromatosis type 1 (NF1)
Children with NF1 have an increased propensity to develop low-grade gliomas, especially in the optic pathway. Up to 20% of patients with NF1 will develop an optic pathway glioma. Most children with NF1-associated optic nerve gliomas are asymptomatic and/or have nonprogressive symptoms and do not require antitumor treatment. Screening magnetic resonance imaging (MRI) in asymptomatic patients with NF1 is usually not indicated, although some investigators perform baseline MRI for young children who cannot undergo detailed ophthalmologic examinations.
The diagnosis is often based on compatible clinical findings and imaging features. Histological confirmation is rarely needed at the time of diagnosis. When biopsies are performed, these tumors are predominantly pilocytic astrocytomas.
Indications for treatment vary, and are often based on the goal of preserving vision.
Very rarely, patients with NF1 develop high-grade gliomas. Sometimes, this tumor is the result of a transformation of a lower-grade tumor.
Tuberous sclerosis
Patients with tuberous sclerosis have a predilection for developing subependymal giant cell astrocytoma (SEGA). Variants in either TSC1 or TSC2 cause constitutive activation of the mammalian target of rapamycin complex 1 (mTORC1) signaling pathway, leading to increases in proliferation. SEGAs are responsive to molecularly targeted approaches with mTORC1 pathway inhibitors.[Level of evidence C2] Patients with tuberous sclerosis are also at risk of developing cortical tubers and subependymal nodules.
Molecular features and recurrent genomic alterations
Recurrent genomic alterations resulting in constitutive activation of the mitogen-activated protein kinase (MAPK) pathway, most commonly involving the BRAF gene, represent the primary (and often sole) oncogenic driver in the vast majority of pediatric low-grade gliomas, including pilocytic/pilomyxoid astrocytomas, gangliogliomas, and others. As a result, most of these tumors are amenable to molecular targeted therapies.
More complex tumor genomes are characteristic of pediatric diffuse high-grade gliomas. These complex genomes include recurrent genomic alterations in the H3 histone encoding genes (e.g., H3F3A, HIST1H3B), DNA damage repair pathways (e.g., TP53, PPM1D, ATM, MDM2), chromatin modifiers (e.g., ATRX, BCOR, SETD2), cell cycle pathways (e.g., CDKN2A, CDKN2B, RB1), and/or oncogene amplifications (PDGFR, VEGFR2, KIT, MYC, MYCN). For most of these tumors, existing conventional and molecular targeted therapies have limited efficacy.
A rare subset of pediatric high-grade gliomas arising in patients with inheritable biallelic mismatch repair deficiency (bMMRD) is characterized by an extraordinarily high mutational burden. Correctly identifying these patients at the time of diagnosis is critical because of intrinsic resistance to temozolomide and responsiveness to treatment with immune checkpoint inhibitors.[Level of evidence C3];
BRAF::KIAA1549
BRAF activation in pilocytic astrocytoma occurs most commonly through a BRAF::KIAA1549 gene fusion, resulting in a fusion protein that lacks the BRAF autoregulatory domain. This fusion is seen in most infratentorial and midline pilocytic astrocytomas, but is present at lower frequency in supratentorial (hemispheric) tumors.
Presence of the BRAF::KIAA1549 fusion is associated with improved clinical outcome (progression-free survival [PFS] and overall survival [OS]) in patients with pilocytic astrocytoma.; [Level of evidence C1] Progression to high-grade gliomas is very rare for pediatric gliomas with the BRAF::KIAA1549 fusion.
BRAF variants
Activating point variants in BRAF, most commonly BRAF V600E, are present in a subset of pediatric gliomas and glioneuronal tumors across a wide spectrum of histologies, including pleomorphic xanthoastrocytoma, pilocytic astrocytoma, ganglioglioma, desmoplastic infantile ganglioglioma/astrocytoma, and others. Some low-grade, infiltrative, pediatric gliomas with an alteration in a MAPK pathway gene, including BRAF, and often resembling diffuse low-grade astrocytoma or oligodendroglioma histologically, are now classified as diffuse low-grade glioma, MAPK pathway altered.
Retrospective clinical studies have shown the following:
- In a retrospective series of more than 400 children with low-grade gliomas, 17% of tumors had BRAF V600E variants. The 10-year PFS rate was 27% for patients with BRAF V600E variants, compared with 60% for patients whose tumors did not harbor that variant. Additional factors associated with this poor prognosis included subtotal resection and CDKN2A deletion.[Level of evidence C2] Even in patients who underwent a gross-total resection, recurrence was noted in one-third, suggesting that BRAF V600E tumors have a more invasive phenotype than do other low-grade glioma variants.
- In a similar analysis, children with diencephalic low-grade astrocytomas with a BRAF V600E variant had a 5-year PFS rate of 22%, compared with a PFS rate of 52% in children with wild-type BRAF.[Level of evidence C2]
- The frequency of the BRAF V600E variant was significantly higher in pediatric low-grade gliomas that transformed to high-grade gliomas (8 of 18 patients) than was the frequency of the variant in tumors that did not transform to high-grade gliomas (10 of 167 cases).
NF1 variants
Somatic alterations in NF1 are seen most frequently in children with NF1 and are associated with germline alterations in the tumor suppressor NF1. Loss of heterozygosity for NF1 represents the most common somatic alteration in these patients followed by inactivating variants in the second NF1 allele, and consistent with a second hit required for tumorigenesis. While most NF1 patients with low-grade gliomas have an excellent long-term prognosis, secondary transformation into high-grade glioma may occur in a small subset. Genomically, transformation is associated with the acquisition of additional oncogenic drivers, such as loss of function alterations in CDKN2A, CDKN2B and/or ATRX. Primary high-grade gliomas may also occur in patients with NF1 but are exceedingly rare. Genomic alterations involving the MAPK signaling pathway other than NF1 are very uncommon in gliomas occurring in children with NF1.
ALK, NTRK1, NTRK2, NTRK3, or ROS1 gene fusions
High-grade gliomas with distinctive molecular characteristics arise in infants, typically in those diagnosed during the first year of life. These tumors are characterized by recurrent oncogenic gene fusions involving ALK, NTRK1, NTRK2, NTRK3, or ROS1 as the primary and, typically, sole oncogenic driver. Infants with this type of glioma, now classified as infant-type hemispheric glioma, have a much better prognosis compared with older children with high-grade gliomas. Remarkably, these tumors may evolve from high-grade to low-grade histology over time, and it remains unclear how much this phenomenon is a consequence of natural disease history versus treatment-induced changes.
ROS1 gene fusions have also been reported in gliomas occurring in older children and adults. A retrospective meta-analysis that included 40 children older than 1 year revealed that ROS1 gene fusions occurred in diverse glioma histologies, including diffuse high-grade and low-grade gliomas and glioneuronal tumors. Similar to ROS1-altered cases occurring in infants, tumor variants in other known driver genes were rare. However, tumor copy number alterations were more frequent in older children than infants.
Other genomic alterations
As an alternative to BRAF activation or NF1 loss, other primary oncogenic driver alterations along the MAPK signaling pathway have been observed in pilocytic astrocytomas and other pediatric-type gliomas. These include oncogenic variants and/or fusions involving FGFR1, FGFR2, PTPN11, RAF1,NTRK2, and others.
Low-grade gliomas with rearrangements in the MYB family of transcription factors have now been classified as a separate entity: diffuse astrocytoma, MYB- or MYBL1-altered, WHO grade 1.
Angiocentric gliomas
Angiocentric gliomas typically arise in children and young adults as cerebral tumors presenting with seizures.
Two reports in 2016 identified MYB gene alterations as being present in almost all cases diagnosed as angiocentric glioma, with QKI being the primary fusion partner in cases where fusion-partner testing was possible. While angiocentric gliomas most commonly occur supratentorially, brain stem angiocentric gliomas with MYB::QKI fusions have also been reported.
Astroblastomas, MN1-altered
Astroblastomas are defined histologically as glial neoplasms composed of GFAP-positive cells and contain astroblastic pseudorosettes that often demonstrate sclerosis. Astroblastomas are diagnosed primarily in childhood through young adulthood.
The following studies have described genomic alterations associated with astroblastoma:
- A report describing a molecular classification of CNS primitive neuroectodermal tumors (PNETs) identified an entity called CNS high-grade neuroepithelial tumor with MN1 alteration (CNS HGNET-MN1) that was characterized by gene fusions involving MN1. Most tumors with a histological diagnosis of astroblastoma (16 of 23) belonged to this molecularly defined entity.
- A report of 27 histologically defined astroblastomas found that 10 cases had MN1 rearrangements, 7 cases had BRAF rearrangements, and 2 cases had RELA rearrangements. Methylation array analysis showed that the cases with MN1 rearrangements clustered with CNS HGNET-MN1, the BRAF-altered cases clustered with pleomorphic xanthoastrocytomas, and the RELA cases clustered with ependymomas.
- Genomic evaluation of eight cases of astroblastoma identified four with MN1 alterations. Of the remaining four cases, two had genomic alterations consistent with high-grade glioma and two cases could not be classified on the basis of their molecular characteristics.
- One study described eight cases of astroblastoma. All five cases that underwent fluorescence in situ hybridization analysis showed MN1 rearrangements.
These reports suggest that the histological diagnosis of astroblastoma encompasses a heterogeneous group of genomically defined entities. Astroblastomas with MN1 fusions represent a distinctive subset of histologically diagnosed cases.
IDH1 and IDH2 variants
IDH1- and IDH2-altered tumors occur in the pediatric population as low-grade gliomas (WHO Grade 2), high-grade gliomas (WHO Grade 3 and 4), and oligodendrogliomas with codeletion of 1p and 19q. For more information about IDH1- and IDH2-altered gliomas, see the IDH1 and IDH2 variants section in the Molecular features of pediatric-type high-grade gliomas section.
Molecular features of pediatric-type high-grade gliomas
Pediatric high-grade gliomas are biologically distinct from those arising in adults.
Subgroups identified using DNA methylation patterns
Pediatric-type high-grade gliomas can be separated into distinct subgroups on the basis of epigenetic patterns (DNA methylation). These subgroups show distinguishing chromosome copy number gains/losses and gene variants in the tumor. Particularly distinctive subtypes of pediatric high-grade gliomas are those with recurring variants at specific amino acids in histone genes, and together these account for approximately one-half of pediatric high-grade gliomas.
The following pediatric-type high-grade glioma subgroups were identified on the basis of their DNA methylation patterns, and they show distinctive molecular and clinical characteristics:
Genomic alterations associated with diffuse midline gliomas
The histone K27 variants: H3.3 (H3F3A) and H3.1 (HIST1H3B and, rarely, HIST1H3C) variants at K27 and EZHIP
The histone K27–altered cases occur predominantly in middle childhood (median age, approximately 10 years), are almost exclusively midline (thalamus, brain stem, and spinal cord), and carry a very poor prognosis. The 2021 WHO classification groups these cancers into a single entity: diffuse midline glioma, H3 K27-altered. However, there are clinical and biological distinctions between cases with H3.3 and H3.1 variants, as described below.
Diffuse midline glioma, H3 K27-altered, is defined by loss of H3 K27 trimethylation either due to an H3 K27M variant or, less commonly, overexpression of EZHIP. This entity includes most high-grade gliomas located in the thalamus, pons (diffuse intrinsic pontine gliomas [DIPGs]), and spinal cord, predominantly in children, but also in adults.
H3.3 K27M: H3.3 K27M cases occur throughout the midline and pons, account for approximately 60% of cases in these locations, and commonly present between the ages of 5 and 10 years. The prognosis for H3.3 K27M patients is especially poor, with a median survival of less than 1 year; the 2-year survival rate is less than 5%. Leptomeningeal dissemination is frequently observed in H3.3 K27M patients.
H3.1 K27M: H3.1 K27M cases are approximately fivefold less common than H3.3 K27M cases. They occur primarily in the pons and present at a younger age than other H3.3 K27M patients (median age, 5 years vs. 6–10 years). These patients have a slightly more favorable prognosis than do H3.3 K27M patients (median survival, 15 months vs. 11 months). Variants in ACVR1, which is also the variant observed in the genetic condition fibrodysplasia ossificans progressiva, are present in a high proportion of H3.1 K27M cases.
H3.2 K27M: Rarely, K27M variants are also identified in H3.2 (HIST2H3C) cases.
A subset of tumors with H3 K27 variants will have a BRAF V600E or FGFR1 co-variant. A retrospective cohort of 29 tumors combined with 31 cases previously reported in the literature demonstrated a somewhat higher propensity for a thalamic location. These cases exhibit a unique DNA methylation cluster that is distinct from other diffuse midline glioma subgroups and glioma subtypes with BRAF or FGFR1 alterations. The median survival for these patients exceeded 3 years. A separate retrospective study of pediatric and adult patients with H3 K27-altered gliomas revealed BRAF V600E variants in 5.8% (9 of 156) and FGFR1 variants in 10.9% (17 of 156) of patients younger than 20 years. Other recurrent genetic alterations detected in pediatric patients included variants in TP53, ATRX, PIK3CA, and amplifications of PDGFRA and KIT. FGFR1 variants were noted to be more frequent in patients older than 20 years (31.8%, 47 of 148).
EZHIP overexpression: The small minority of patients with diffuse midline gliomas lacking histone H3 variants often show EZHIP overexpression. EZHIP inhibits PRC2 activity, leading to the same loss of H3 K27 trimethylation that is induced by H3 K27M variants. Overexpression of EZHIP is likewise observed in posterior fossa type A ependymomas, which also shows loss of H3 K27 methylation.
H3.3 (H3F3A) variant at G34
The H3.3 G34 subtype arises from H3.3 glycine 34 to arginine/valine (G34R/V) variants. This subtype presents in older children and young adults (median age, 14–18 years) and arises exclusively in the cerebral cortex. H3.3 G34 cases commonly have variants in TP53 and ATRX (95% and 84% of cases, respectively, in one large series) and show widespread hypomethylation across the whole genome. In a series of 95 patients with the H3.3 G34 subtype, 44% of patients also had a variant in PDGFRA at the time of diagnosis, and 81% of patients had PDGFRA variants observed at relapse.
Patients with H3F3A variants are at high risk of treatment failure, but the prognosis is not as poor as that of patients with histone 3.1 or 3.3 K27M variants. O-6-methylguanine-DNA methyltransferase (MGMT) methylation is observed in approximately two-thirds of cases, and aside from the IDH1-altered subtype (see below), the H3.3 G34 subtype is the only pediatric high-grade glioma subtype that demonstrates MGMT methylation rates exceeding 20%.
IDH1 and IDH2 variants
IDH1- and IDH2-altered tumors occur in the pediatric population as low-grade gliomas (WHO grade 2), high-grade gliomas (WHO grades 3 and 4), and oligodendrogliomas with codeletion of 1p and 19q.
- IDH1 variants are much more common than IDH2 variants, accounting for approximately 90% of pediatric IDH-altered CNS tumors.
- IDH-altered low-grade gliomas are more common than IDH-altered high-grade gliomas, accounting for approximately three-fourths of IDH-altered pediatric glioma cases.
- Oligodendrogliomas with IDH variants represent approximately 20% of pediatric CNS tumors with IDH variants.
- The median age at diagnosis for pediatric patients with IDH-altered tumors is approximately 16 years, and IDH-altered CNS tumors are very uncommon in children aged 10 years and younger.
- Like astrocytomas with IDH variants in adults, those in affected children commonly have TP53 variants (approximately 90% of cases) and ATRX variants (approximately 50%).
- Like IDH-altered, low-grade gliomas in adults, low-grade tumors in pediatric patients can also show progression to high-grade gliomas.
IDH1-altered cases represent a small percentage of high-grade gliomas (approximately 5%–10%) seen in pediatrics, and are almost exclusively older adolescents (median age in a pediatric population, 16 years) with hemispheric tumors. These tumors are classified under adult-type diffuse glioma, as astrocytoma, IDH-altered in the 2021 WHO CNS classification. IDH1-altered cases often show TP53 variants, MGMT promoter methylation, and a glioma-CpG island methylator phenotype (G-CIMP).
Pediatric patients with IDH1 variants have a more favorable prognosis than patients with other types of high-grade gliomas. A retrospective multi-institutional review of pediatric patients with IDH-altered gliomas and available outcome data (n = 76) reported a 5-year PFS rate of 44% (95% CI, 25%–59%) and a 5-year OS rate of 92% (95% CI, 79%–97%). Approximately 25% of the gliomas in the cohort were classified as high grade. There was no difference in 5-year PFS rates observed between tumor grades. However, patients with high-grade tumors had a worse 5-year OS rate of 75% (95% CI, 40%–91%).
Rare, IDH-altered, high-grade gliomas have been reported to occur in children with mismatch repair–deficiency syndromes (Lynch syndrome or constitutional mismatch repair deficiency syndrome). These tumors, termed primary mismatch repair–deficient IDH-altered astrocytomas (PMMRDIAs), could be distinguished from other IDH-altered gliomas by methylation profiling. PMMRDIAs have molecular features that are distinct from most IDH-altered gliomas, including a hypervariant phenotype and frequent activation of receptor tyrosine kinase pathways. Patients with PMMRDIAs have a markedly worse prognosis than patients with other IDH-altered gliomas, with a median survival of 15 months.
Pleomorphic xanthoastrocytoma (PXA)–like
Approximately 10% of pediatric high-grade gliomas have DNA methylation patterns that are PXA-like. PXA-like cases commonly have BRAF V600E variants and a relatively favorable outcome (approximately 50% survival at 5 years).
High-grade astrocytoma with piloid features
This entity was included in the 2016 WHO classification (called pilocytic astrocytoma with anaplasia) to describe tumors with histological features of pilocytic astrocytoma, increased mitotic activity, and additional high-grade features. The current nomenclature was adopted in the 2021 WHO classification. A more recent publication described a cohort of 83 cases with these histological features (referred to as anaplastic astrocytoma with piloid features) that shared a common DNA methylation profile, which is distinct from the methylation profiles of other gliomas. These tumors occurred more often in adults (median age, 41 years), and they harbored frequent deletions of CDKN2A/B, MAPK pathway alterations (most often in the NF1 gene), and variants or deletions of ATRX. They are associated with a clinical course that is intermediate between pilocytic astrocytoma and IDH–wild-type glioblastoma.
Other variants
Pediatric patients with glioblastoma multiforme high-grade glioma whose tumors lack both histone variants and IDH1 variants represent approximately 40% of pediatric glioblastoma multiforme cases. This is a heterogeneous group, with higher rates of gene amplifications than other pediatric high-grade glioma subtypes. The most commonly amplified genes are PDGFRA, EGFR, CCND/CDK, and MYC/MYCN. MGMT promoter methylation rates are low in this group. One report divided this group into three subtypes. The subtype characterized by high rates of MYCN amplification showed the poorest prognosis, while the subtype characterized by TERT promoter variants and EGFR amplification showed the most favorable prognosis. The third group was characterized by PDGFRA amplification.
High-grade gliomas in infants
Infants and young children with high-grade gliomas appear to have tumors with distinctive molecular characteristics when compared with tumors of older children and adults with high-grade gliomas. An indication of this difference was noted with the application of DNA methylation analysis to pediatric high-grade tumors, which found that approximately 7% of pediatric patients with a histological diagnosis of high-grade glioma had tumors with methylation patterns more closely resembling those of low-grade gliomas. Ten of 16 infants (younger than 1 year) with a high-grade glioma diagnosis were in this methylation array–defined group. The 5-year survival rate for patients in this report diagnosed at younger than 1 year exceeded 60%, while the 5-year survival rate for patients aged 1 to 3 years and older was less than 20%.
Two studies of the molecular characteristics of high-grade gliomas in infants and young children have further defined the distinctive nature of tumors arising in children younger than 1 year. A key finding from both studies is the importance of gene fusions involving tyrosine kinases (e.g., ALK, NTRK1, NTRK2, NTRK3, and ROS1) in patients in this age group. Both studies also found that infants with high-grade gliomas whose tumors have these gene fusions have survival rates much higher than those of older children with high-grade gliomas.
The first study presented data for 118 children younger than 1 year with a low-grade or high-grade glioma diagnosis who had tumor tissue available for genomic characterization. Approximately 75% of the cases were classified as low grade, but the diminished utility of histological classification in this age group was illustrated by the relatively low OS rate for the low-grade cohort (71%) and the relatively favorable survival for the high-grade cohort (55%). Rates of surgical resection were higher for patients with high-grade tumors, a result of many of the low-grade tumors occurring in midline locations while the high-grade tumors were found in supratentorial locations. This finding may also help to explain the relative outcomes for the two groups. Genomic characterization divided the infant glioma population into the following three groups, the first of which included patients with high-grade gliomas:
- Group 1 tumors were receptor tyrosine kinase driven and primarily high grade (83%). These tumors harbored lesions in ALK, ROS1, NTRK, and MET. The median age at diagnosis was 3 months, and OS rates were approximately 60%.
- Group 2 tumors were RAS/MAPK driven and were all hemispheric low-grade gliomas, representing one-fourth of hemispheric gliomas in infants. BRAF V600E was the most common alteration, followed by FGFR1 alterations and BRAF fusions. This group had a median age at presentation of 8 months and had the most favorable outcome (10-year OS rate, 93%).
- Group 3 tumors were RAS/MAPK driven with low-grade histology and midline presentation (approximately 80% optic pathway/hypothalamic gliomas). Most group 3 tumors showed either BRAF fusions or BRAF V600E. Median age at diagnosis was 7.5 months. The 5-year progression-free survival (PFS) rate was approximately 20%, and the 10-year OS rate was approximately 50% (far inferior to that of optic pathway/hypothalamic gliomas in children aged >1 year).
The second study focused on tumors from children younger than 4 years with a pathological diagnosis of WHO grades 2, 3, and 4 gliomas, astrocytomas, or glioneuronal tumors. Among the 191 tumors studied that met inclusion criteria, 61 had methylation profiles consistent with glioma subtypes that occur in older children (e.g., IDH1, diffuse midline glioma H3 K27-altered, SEGA, pleomorphic xanthoastrocytoma, etc.). The remaining 130 cases were called the intrinsic set and were the focus of additional molecular characterization:
- The intrinsic set contained most of the patients diagnosed before age 1 year (49 of 63 patients, 78%) and had a median age of 7.2 months. Tumors were frequently in a superficial hemispheric location, often involving the meninges, and had a well-defined border with adjacent normal brain.
- The methylation classifier placed most of these cases in either the desmoplastic infantile ganglioglioma/astrocytoma (DIG/DIA) subgroup or in the infantile hemispheric glioma subgroup.
- For 41 tumors from the intrinsic set in which tissue was available for gene panel and RNA sequencing, 25 tumors had fusions involving either ALK (n = 10), NTRK1 (n = 2), NTRK2 (n = 2), NTRK3 (n = 8), ROS1 (n = 2), or MET (n = 1). BRAF variants (n = 3) were observed in cases that were high scoring by methylation array for the DIG/DIA or DIG/DIA-like subgroups.
- For patients in the intrinsic set, the 5-year survival rate was higher for patients whose tumors had gene fusions when compared with patients whose tumors lacked fusions (approximately 80% vs. 60%, respectively). However, both of these groups of patients had much higher survival rates than other children with high-grade gliomas.
Secondary high-grade glioma
Childhood secondary high-grade glioma (high-grade glioma that is preceded by a low-grade glioma) is uncommon (2.9% in a study of 886 patients). No pediatric low-grade gliomas with the BRAF::KIAA1549 fusion transformed to a high-grade glioma, whereas low-grade gliomas with the BRAF V600E variants were associated with increased risk of transformation. Seven of 18 patients (approximately 40%) with secondary high-grade glioma had BRAF V600E variants, with CDKN2A alterations present in 8 of 14 cases (57%).
Molecular features of glioneuronal and neuronal tumors
Glioneuronal and neuronal tumors are generally low-grade tumors. Select histologies recognized by the 2021 WHO classification include the following:
- Ganglioglioma.
- Desmoplastic infantile ganglioglioma/desmoplastic infantile astrocytoma.
- Dysembryoplastic neuroepithelial tumor.
- Papillary glioneuronal tumor.
- Rosette-forming glioneuronal tumor.
- Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease).
- Gangliocytoma.
- Diffuse leptomeningeal glioneuronal tumor.
- Central neurocytoma.
- Extraventricular neurocytoma.
Ganglioglioma
Ganglioglioma presents during childhood and into adulthood. It most commonly arises in the cerebral cortex and is associated with seizures, but it also presents in other sites, including the spinal cord.
The unifying theme for the molecular pathogenesis of ganglioglioma is genomic alterations leading to MAPK pathway activation. BRAF alterations are observed in approximately 50% of ganglioglioma cases, with V600E being by far the most common alteration. However, other BRAF variants and gene fusions are also observed. Other less commonly altered genes in ganglioglioma include KRAS, FGFR1, FGFR2, RAF1, NTRK2, and NF1.
Desmoplastic infantile astrocytomas (DIA) and desmoplastic infantile gangliogliomas (DIG)
DIA and DIG most often present in the first year of life and show a characteristic imaging appearance in which a contrast-enhancing solid nodule accompanies a large cystic component. DIG is more common than DIA, and by methylation array analysis, both diagnoses cluster together. Survival outcome is generally favorable with surgical resection.
The most commonly observed genomic alterations in DIA and DIG are BRAF variants involving V600. Gene fusions involving kinase genes are observed less frequently.
- Among 16 cases confirmed by histology and DNA methylation profiling to be DIA and DIG, BRAF variants were observed in seven cases (43.8%): four BRAF V600E variants and three BRAF V600D variants. One additional case had an EML4::ALK fusion. BRAF variants were present in 4 of 12 DIG cases (25%) (with 3 of 4 altered cases having BRAF V600D) and in 3 of 4 DIA cases (75%) (all 3 altered cases with BRAF V600E).
- One study of seven DIG cases found MAPK pathway alterations in four (57%). Three alterations involved BRAF (V600E, V600D, and one deletion/insertion centered at V600) and one was a TPM3::NTRK1 in-frame fusion. Notably, the variant allele frequency was low (8%–27%), suggesting that DIG is characterized by a prominent nonneoplastic component resulting in low clonal driver variant allele frequencies.
- Another report also described the BRAF V600D variant in a DIG case. As the V600D variant is far less common than V600E in other cancers, its detection in multiple DIG cases suggests an association between the variant and DIG.
Dysembryoplastic neuroepithelial tumor (DNET)
DNET presents in children and adults, with the median age at diagnosis in mid-to-late adolescence. It is characterized histopathologically by the presence of columns of oligodendroglial-like cells and cortical ganglion cells floating in mucin. The temporal lobe is the most common location, and it is associated with drug-refractory epilepsy.
FGFR1 alterations have been reported in 60% to 80% of DNETs, and include FGFR1 activating point variants, internal tandem duplication of the kinase domain, and activating gene fusions. BRAF variants are uncommon in DNET.
Papillary glioneuronal tumor
Papillary glioneuronal tumor is a low-grade biphasic neoplasm with astrocytic and neuronal differentiation that primarily arises in the supratentorial compartment. The median age at presentation is in the early 20s, but it can be observed during childhood through adulthood.
The primary genomic alteration associated with papillary glioneuronal tumor is a gene fusion, SLC44A1::PRKCA, that is associated with the t(9:17)(q31;q24) translocation. In one study of 28 cases diagnosed histologically as papillary glioneuronal tumor using methylation arrays, 11 of the cases clustered in a distinctive methylation class, while the remaining cases showed methylation profiles typical for other tumor entities. Molecular analysis of the cases in the distinctive methylation cluster showed that all of them had the SLC44A1::PRKCA gene fusion except for a single case with a NOTCH1::PRKCA gene fusion. This suggests that molecular methods for identifying the presence of a PRKCA fusion are less susceptible to misclassification in diagnosing papillary glioneuronal tumor than are morphology-based methods.
Rosette-forming glioneuronal tumor (RGNT)
RGNT presents in adolescents and adults, with tumors generally located infratentorially, although tumors can arise in mesencephalic or diencephalic regions. The typical histological appearance shows both a glial component and a neurocytic component arranged in rosettes or perivascular pseudorosettes. Outcome for patients with RGNT is generally favorable, consistent with the WHO grade 1 designation.
DNA methylation profiling shows that RGNT has a distinct epigenetic profile that distinguishes it from other low-grade glial/glioneuronal tumor entities. A study of 30 cases of RGNT observed FGFR1 hotspot variants in all analyzed tumors. In addition, PIK3CA activating variants were concurrently observed in 19 of 30 cases (63%). Missense or damaging variants in NF1 were identified in 10 of 30 cases (33%), with 7 tumors having variants in FGFR1, PIK3CA, and NF1. The co-occurrence of variants that activate both the MAPK pathway and the PI3K pathway makes the variant profile of RGNT distinctive among astrocytic and glioneuronal tumors.
Diffuse leptomeningeal glioneuronal tumor (DLGNT)
DLGNT is a rare CNS tumor that has been characterized radiographically by leptomeningeal enhancement on MRI that may involve the posterior fossa, brain stem region, and spinal cord. Intraparenchymal lesions, when present, typically involve the spinal cord. Localized intramedullary glioneuronal tumors without leptomeningeal dissemination and with histomorphological, immunophenotypic, and genomic characteristics similar to DLGNT have been reported.
DLGNT showed a distinctive epigenetic profile on DNA methylation arrays, and unsupervised clustering of array data applied to 30 cases defined two subclasses of DLGNT: methylation class (MC)-1 (n = 17) and MC-2 (n = 13). Of note, many of the array-defined cases had originally been diagnosed as other entities (e.g., primitive neuroectodermal tumors, pilocytic astrocytoma, and anaplastic astrocytoma). Patients with DLGNT-MC-1 were diagnosed at an earlier age than were patients with DLGNT-MC-2 (5 years vs. 14 years, respectively). The 5-year OS rate was higher for patients with DLGNT-MC-1 than for those with DLGNT-MC-2 (100% vs. 43%, respectively). Genomic findings from the 30 cases of methylation array–defined DLGNT are provided below:
- All 30 cases showed loss of chromosome 1p, but only 6 of 17 DLGNT-MC-1 cases showed additional gain of chromosome 1q, compared with all cases of DLGNT-MC-2. A separate report found that chromosome 1q gain was an adverse prognostic factor in patients with DLGNT (including cases with localized disease), which is consistent with the inferior outcome for patients with DLGNT-MC-2.
- Co-deletions of 1p/19q were more frequent in the DLGNT-MC-1 group (7 of 13, 54%) than in the DLGNT-MC-2 group (2 of 13, 15%). In contrast to oligodendroglioma, variants of IDH1 and IDH2 were not identified.
- MAPK pathway activation is common in DLGNT cases. The KIAA1549::BRAF fusion was present in 11 of 15 DLGNT-MC-1 cases (65%) and in 9 of 13 DLGNT-MC-2 cases (69%). Fusions involving NTRK1, NTRK2, or NTRK3 were present in one case each, and another case had a TRIM33::RAF1 fusion.
Extraventricular neurocytoma
Extraventricular neurocytoma is histologically similar to central neurocytoma, consisting of small uniform cells that demonstrate neuronal differentiation. However, extraventricular neurocytoma arises in the brain parenchyma rather than in association with the ventricular system. It presents during childhood through adulthood.
In a study of 40 tumors histologically classified as extraventricular neurocytoma and subjected to methylation array analysis, only 26 formed a separate cluster distinctive from reference tumors of other histologies. Among cases with an extraventricular neurocytoma methylation array classification for which genomic characterization could be performed, 11 of 15 (73%) showed rearrangements affecting members of the FGFR family, with FGFR1::TACC1 being the most common alteration.
For information about the treatment of gliomas, glioneuronal tumors, and neuronal tumors, see Childhood Astrocytomas, Other Gliomas, and Glioneuronal/Neuronal Tumors Treatment.
Central Nervous System (CNS) Atypical Teratoid/Rhabdoid Tumors (AT/RT)
SMARCB1 and SMARCA4 genes
AT/RT was the first primary pediatric brain tumor in which a candidate tumor suppressor gene, SMARCB1, was identified. SMARCB1 is genomically altered in most rhabdoid tumors, including CNS, renal, and extrarenal rhabdoid malignancies. SMARCB1 is a component of the SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin-remodeling complex.
Rare cases of rhabdoid tumors expressing SMARCB1 and lacking SMARCB1 variants have also been associated with somatic or germline variants of SMARCA4, another member of the SWI/SNF chromatin-remodeling complex.
Less commonly, SMARCA4-negative (with retained SMARCB1) tumors have been described. Loss of SMARCB1 or SMARCA4 staining is a defining marker for AT/RT.
The 2021 WHO classification defines AT/RT by the presence of either SMARCB1 or SMARCA4 alterations. Tumors with histological features of AT/RT that lack these genomic alterations are termed CNS embryonal tumors with rhabdoid features.
Despite the absence of recurring genomic alterations beyond SMARCB1 and SMARCA4, biologically, relatively distinctive subsets of AT/RT have been identified. In one study, three distinctive subsets of AT/RT were identified through the use of DNA methylation arrays for 150 AT/RT tumors and gene expression arrays for 67 AT/RT tumors:
- AT/RT tyrosinase (TYR): This subset represented approximately one-third of cases and was characterized by elevated expression of melanosomal markers such as TYR (the gene encoding tyrosinase). Cases in this subset were primarily infratentorial, with most presenting in children aged 0 to 1 year and showing chromosome 22q loss. For patients with AT/RT TYR, the mean overall survival (OS) was 37 months in a clinically heterogeneous group (95% confidence interval [CI], 18–56 months). In the prospective European Rhabdoid Registry (EU-RHAB) series, patients aged 1 year and older with AT/RT TYR demonstrated a 5-year OS rate of 71%, while those younger than 1 year with a non-TYR AT/RT had a very poor survival rate.
- AT/RT sonic hedgehog (SHH): This subset represented approximately 40% of cases and was characterized by elevated expression of genes in the SHH pathway (e.g., GLI2 and MYCN). Cases in this subset occurred with similar frequency in the supratentorium and infratentorium. While most patients presented before the age of 2 years, approximately one-third of patients presented between the ages of 2 and 5 years. For patients with AT/RT SHH, the mean OS was 16 months (95% CI, 8–25 months).
In a subsequent study, the AT/RT SHH subgroup was further divided into three subtypes: SHH-1A, SHH-1B, and SHH-2. Children older than 3 years who harbored the SHH-1B signature experienced the most favorable outcomes.
- AT/RT MYC: This subset represented approximately one-fourth of cases and was characterized by elevated expression of MYC. AT/RT MYC cases tended to occur in the supratentorial compartment. While most AT/RT MYC cases occurred by the age of 5 years, AT/RT MYC represented the most common subset diagnosed at age 6 years and older. Focal deletions of SMARCB1 were the most common mechanism of SMARCB1 loss for this subset. For patients with AT/RT MYC, the mean OS was 13 months (95% CI, 5–22 months).
Loss of SMARCB1 or SMARCA4 protein expression has therapeutic significance, because this loss creates a dependence of the cancer cells on EZH2 activity. Preclinical studies have shown that some AT/RT xenograft lines with SMARCB1 loss respond to EZH2 inhibitors with tumor growth inhibition and occasional tumor regression. In a study of the EZH2 inhibitor tazemetostat, objective responses were observed in adult patients whose tumors had either SMARCB1 or SMARCA4 loss (non-CNS malignant rhabdoid tumors and epithelioid sarcoma). For more information, see the Treatment of Recurrent Childhood CNS Atypical Teratoid/Rhabdoid Tumor section.
For information about the treatment of childhood CNS AT/RTs, see Childhood Central Nervous System Atypical Teratoid/Rhabdoid Tumors Treatment.
Medulloblastomas
Molecular subtypes of medulloblastoma
Multiple medulloblastoma subtypes have been identified by integrative molecular analysis. Since 2012, the general consensus is that medulloblastoma can be molecularly separated into at least four core subtypes, including WNT-activated, sonic hedgehog (SHH)–activated, group 3, and group 4. In the 2021 World Health Organization (WHO) classification, the SHH subgroup has been divided into two groups on the basis of TP53 status. Group 3 and group 4, which require methylation analysis for reliable separation, have been combined into medulloblastoma, non-WNT/non-SHH. Because the group 3 and group 4 terminology has been used extensively in completed studies and is still in use in ongoing and planned studies, this nomenclature will be maintained throughout the clinical discussion in this summary.
Different regions of the same tumor are likely to have other disparate genetic mutations, adding to the complexity of devising effective molecularly targeted therapy. However, the major subtypes noted above remain stable across primary and metastatic components.
Further subclassification within these subgroups is possible, which will provide even more prognostic information.
Medulloblastoma, WNT-activated
WNT tumors are medulloblastomas with aberrations in the WNT signaling pathway and represent approximately 10% of all medulloblastomas. WNT medulloblastomas show a WNT signaling gene expression signature and beta-catenin nuclear staining by immunohistochemistry. They are usually histologically classified as classic medulloblastoma tumors and rarely have a large cell/anaplastic appearance. WNT medulloblastomas generally occur in older patients (median age, 10 years) and are infrequently metastasized at diagnosis. Recent studies have demonstrated the value of methylation profiling in identifying WNT-activated medulloblastomas. These studies included cases that would not be detected using other current testing methods (e.g., beta-catenin immunohistochemistry, CTNNB1 mutation analysis, and evaluation for monosomy 6).
CTNNB1 mutations are observed in 85% to 90% of WNT medulloblastoma cases, with APC mutations detected in many of the cases that lack CTNNB1 mutations. Patients with WNT medulloblastoma whose tumors have APC mutations often have Turcot syndrome (i.e., germline APC mutations). In addition to CTNNB1 mutations, WNT medulloblastoma tumors show 6q loss (monosomy 6) in 80% to 90% of cases. While monosomy 6 is observed in most medulloblastoma patients younger than 18 years at diagnosis, it appears to be much less common (approximately 25% of cases) in patients older than 18 years.
The WNT subset is primarily observed in older children, adolescents, and adults and does not show a male predominance. The subset is believed to have brain stem origin, from the embryonal rhombic lip region. WNT medulloblastomas are associated with a very good outcome in children, especially in individuals whose tumors have beta-catenin nuclear staining and proven 6q loss and/or CTNNB1 mutations. Retrospective studies have suggested that additional TP53 mutations and OTX2 copy number gains may be associated with a worse prognosis for patients with WNT medulloblastoma.
Medulloblastoma, SHH-activated and TP53-mutant and medulloblastoma, SHH-activated and TP53-wild type
SHH tumors are medulloblastomas with aberrations in the SHH pathway and represent approximately 25% of medulloblastoma cases. SHH medulloblastomas are characterized by chromosome 9q deletions; desmoplastic/nodular histology; and mutations in SHH pathway genes, including PTCH1, PTCH2, SMO, SUFU, and GLI2.
Heterozygous deleterious germline mutations in the GPR161 gene were identified in approximately 3% of cases of SHH medulloblastoma. GPR161 is an inhibitor of SHH signaling. Median age at diagnosis for GPR161-mutated cases was 1.5 years. Loss of heterozygosity (LOH) at the GPR161 locus was noted in all tumors, with tumors from five of six patients showing copy-neutral LOH of chromosome 1q (on which GPR161 resides).
Mutations in the third nucleotide (r.3A>G) of the U1 spliceosomal small nuclear RNAs (snRNAs) are highly specific for SHH medulloblastoma. U1 snRNA r.3A>G mutations are observed in virtually all cases of SHH medulloblastoma in adults, in approximately one-third of cases in children and adolescents, and are absent in infant cases. U1 snRNA mutations disrupt RNA splicing, leading to inactivation of tumor-suppressor genes (e.g., PTCH1) and activation of oncogenes (e.g., GLI2). The significance of U1 snRNA r.3A>G mutations in specific SHH medulloblastoma subtypes is described below.
SHH medulloblastomas show a bimodal age distribution and are observed primarily in children younger than 3 years and in older adolescence/adulthood. The tumors are believed to emanate from the external granular layer of the cerebellum. The heterogeneity in age at presentation maps to distinctive subsets identified by further molecular characterization, as follows:
- The subset of medulloblastoma most common in children aged 3 to 16 years, termed SHH-alpha, is enriched for MYCN and GLI2 amplifications, with TP53 mutations commonly co-occurring with one of these amplifications. PTCH1 mutations occur in this subtype and are mutually exclusive with TP53 mutations (often germline), while SMO and SUFU mutations are rare. U1 snRNA mutations occur in approximately 25% of SHH-alpha medulloblastoma cases and are associated with a very poor prognosis.
- Two SHH subtypes that occur primarily in children younger than 3 years have been described. One of these subtypes, termed SHH-beta, is more frequently metastatic, with more frequent focal amplifications. The second of these subtypes, termed SHH-gamma, is enriched for the medulloblastoma with extensive nodularity (MBEN) histology. SHH pathway mutations in children younger than 3 years with medulloblastoma include PTCH1 and SUFU mutations. SUFU mutations are rarely observed in older children and adults, and they are commonly germline events.
Reports that used DNA methylation arrays have also identified two subtypes of SHH medulloblastoma in young children. One of the subtypes contained all of the cases with SMO mutations, and it was associated with a favorable prognosis. The other subtype had most of the SUFU mutations, and it was associated with a much lower progression-free survival (PFS) rate. PTCH1 mutations were present in both subtypes.
- A fourth SHH subtype, termed SHH-delta, includes most of the adult cases of SHH medulloblastoma. Virtually all cases of SHH-delta medulloblastoma have the U1 snRNA r.A>3 mutation, and approximately 90% of cases have TERT promoter mutations. PTCH1 and SMO mutations are also observed in adults with SHH medulloblastoma.
The outcome for patients with nonmetastatic SHH medulloblastoma is relatively favorable for children younger than 3 years and for adults. Young children with the MBEN histology have a particularly favorable prognosis. Patients with SHH medulloblastoma at greatest risk of treatment failure are children older than 3 years whose tumors have TP53 mutations, often with co-occurring GLI2 or MYCN amplification and large cell/anaplastic histology.
Patients with unfavorable molecular findings have an unfavorable prognosis, with fewer than 50% of patients surviving after conventional treatment.
The 2021 WHO classification identifies SHH medulloblastoma with a TP53 mutation as a distinctive entity (medulloblastoma, SHH-activated and TP53-mutant). Approximately 25% of SHH-activated medulloblastoma cases have TP53 mutations, with a high percentage of these cases also showing a TP53 germline mutation (9 of 20 in one study). These patients are commonly between the ages of 5 years and 18 years and have a worse outcome (overall survival at 5 years, <50%). The tumors often show large cell anaplastic histology.
Medulloblastoma, non–WNT/non–SHH-activated
The WHO classification combines group 3 and group 4 medulloblastoma cases into a single entity, partly on the basis of the absence of immediate clinical impact for this distinction. Group 3 medulloblastoma represents approximately 25% of medulloblastoma cases, while group 4 medulloblastoma represents approximately 40% of medulloblastoma cases. Both group 3 and group 4 medulloblastoma patients are predominantly male. Group 3 and group 4 medulloblastomas can be further subdivided on the basis of characteristics such as gene expression and DNA methylation profiles, but the optimal approach to their subdivision is not established.
Various genomic alterations are observed in group 3 and group 4 medulloblastomas; however, no single alteration occurs in more than 10% to 20% of cases. Genomic alterations include the following:
- MYC amplification was the most common distinctive alteration reported for group 3 medulloblastoma, occurring in approximately 15% of cases.
- The most common distinctive genomic alteration described for group 4 medulloblastoma (observed in approximately 15% of cases) was activation of PRDM6 by enhancer hijacking, resulting from the tandem duplication of the adjacent SNCAIP gene.
- Other genomic alterations were observed in both group 3 and group 4 cases, including MYCN amplification and structural variants leading to GFI1 or GFI1B overexpression through enhancer hijacking.
- Isochromosome 17q (i17q) is the most common cytogenetic abnormality and is observed in a high percentage of group 4 cases, as well as in group 3 cases, but it is rarely observed in WNT and SHH medulloblastoma. Prognosis for group 3 and group 4 patients does not appear to be affected by the presence of i17q.
Group 3 patients with MYC amplification or MYC overexpression have a poor prognosis. Fewer than 50% of these patients survive 5 years after diagnosis. This poor prognosis is especially true in children younger than 4 years at diagnosis. However, patients with group 3 medulloblastoma without MYC amplification who are older than 3 years have a prognosis similar to that of most patients with non-WNT medulloblastoma, with a 5-year PFS rate higher than 70%.
Group 4 medulloblastomas occur throughout infancy and childhood and into adulthood. The prognosis for group 4 medulloblastoma patients is similar to that for patients with other non-WNT medulloblastomas and may be affected by additional factors such as the presence of metastatic disease, chromosome 11q loss, and chromosome 17p loss. One study found that group 4 patients with either chromosome 11 loss or gain of chromosome 17 were low risk, regardless of metastases. In cases lacking both of these cytogenetic features, metastasis at presentation differentiated between high and intermediate risk.
For group 3 and group 4 standard-risk patients (i.e., without MYC amplification or metastatic disease), the gain or loss of whole chromosomes appears to connote a favorable prognosis. This finding was derived from the data of 91 patients with non-WNT/non-SHH medulloblastoma enrolled in the SIOP-PNET-4 (NCT01351870) clinical trial and was confirmed in an independent group of 70 children with non-WNT/non-SHH medulloblastoma treated between 1990 and 2014. Chromosomal abnormalities include the following:
- The gain/loss of one or more whole chromosomes was associated with a 5-year event-free survival (EFS) rate of 93%, compared with 64% for no whole chromosome gains/losses.
- The most common whole chromosomal gains/losses are gain of chromosome 7 and loss of chromosomes 8 and 11.
- The optimally performing prognosis discriminator was determined to be the occurrence of two or more of the following aberrations: chromosome 7 gain, chromosome 8 loss, and chromosome 11 loss. Approximately 40% of group 3 and group 4 standard-risk patients had two or more of these chromosomal aberrations and had a 5-year EFS rate of 100%, compared with 68% for patients with fewer than two aberrations.
- In an independent cohort, the prognostic significance of two or more gains/losses versus zero or one gain/loss of chromosomes 7, 8, and 11 was confirmed (5-year EFS rate, 95% for patients with two or more vs. 59% for patients with one or fewer).
The classification of medulloblastoma into the four major subtypes will likely be altered in the future. Further subdivision within subgroups based on molecular characteristics is likely because each of the subgroups is further molecularly dissected, although the studies are nearing consensus as data from multiple independent studies are merged. As an example, using complementary bioinformatics approaches, concordance was analyzed between multiple large published cohorts, and a more unified subgrouping was described. For children with group 3 and group 4 medulloblastomas, eight distinct subgroups were determined by DNA methylation clustering. Specific subgroups had different prognoses.
It is unknown whether the classification for adults with medulloblastoma has a predictive ability similar to that for children. In one study of adult medulloblastoma, MYC oncogene amplifications were rarely observed, and tumors with 6q deletion and WNT activation (as identified by nuclear beta-catenin staining) did not share the excellent prognosis seen in pediatric medulloblastomas, although another study did confirm an excellent prognosis for WNT-activated tumors in adults.
For information about the treatment of childhood medulloblastoma, see Childhood Medulloblastoma and Other Central Nervous System Embryonal Tumors Treatment.
Nonmedulloblastoma Embryonal Tumors
This section describes the genomic characteristics of embryonal tumors other than medulloblastoma and atypical teratoid/rhabdoid tumor. The 2016 WHO classification removed the term primitive neuroectodermal tumors (PNET) from the diagnostic lexicon. This change resulted from the recognition that many tumors previously classified as CNS PNETs have the common finding of amplification of the C19MC region on chromosome 19. These entities included ependymoblastoma, embryonal tumors with abundant neuropil and true rosettes (ETANTR), and some cases of medulloepithelioma. The 2016 WHO classification categorized tumors with C19MC amplification as embryonal tumor with multilayered rosettes (ETMR), C19MC-altered. Tumors previously classified as CNS PNETs are now termed CNS embryonal tumor, not otherwise specified (NOS), with the recognition that tumors in this category will likely be classified by their defining genomic lesions in future editions of the WHO classification.
The 2021 WHO classification categorizes nonmedulloblastoma embryonal tumors primarily by histological and immunohistological features and, in some cases, by molecular findings. The classification includes the following:
- AT/RT.
- Cribriform neuroepithelial tumor.
- ETMR.
- CNS neuroblastoma, FOXR2-activated.
- CNS tumor with BCOR internal tandem duplication.
- CNS embryonal tumor, not elsewhere classified (NEC)/NOS.
NEC is defined as a tumor not elsewhere classified. The NOS nomenclature is used for tumors that cannot be further classified. For many lesions, there are overlapping histological features, and methylation-based clustering is critical for specific diagnosis.
Molecular subtypes of nonmedulloblastoma embryonal tumors
Studies applying unsupervised clustering of DNA methylation patterns for nonmedulloblastoma embryonal tumors found that approximately one-half of these tumors diagnosed as nonmedulloblastoma embryonal tumors showed molecular profiles characteristic of other known pediatric brain tumors (e.g., high-grade glioma). These observations highlight the utility of molecular characterization to assign this class of tumors to their appropriate biology-based diagnosis.
Among the tumors diagnosed as nonmedulloblastoma embryonal tumors, molecular characterization identified genomically and biologically distinctive subtypes, including the following:
- Cribriform neuroepithelial tumor: Representing less than 50% of nonmedulloblastoma embryonal tumors, this subtype is a nonrhabdoid tumor that arises in the vicinity of the third, fourth, or lateral ventricles. This tumor is characterized by cribriform strands and ribbons and demonstrates loss of nuclear SMARCB1 expression. The median age at diagnosis is 20 months. This tumor occurs more often in males, with a male-to-female ratio of 1.5 to 1.
- Embryonal tumor with multilayered rosettes (ETMR): Representing up to 20% of nonmedulloblastoma embryonal tumors, this subtype combines embryonal, rosette-forming, neuroepithelial brain tumors that were previously categorized as either embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, or medulloepithelioma. ETMRs arise most commonly in young children (median age at diagnosis, 2–3 years) but may occur in older children.
ETMRs are defined at the molecular level by high-level amplification of the microRNA cluster C19MC and by a gene fusion between TTYH1 and C19MC. This gene fusion puts expression of C19MC under control of the TTYH1 promoter, leading to high-level aberrant expression of the microRNAs within the cluster. The World Health Organization (WHO) allows histologically similar tumors without C19MC alteration to be classified as ETMR, not otherwise specified (NOS). This subclass of tumors without C19MC alterations may harbor biallelic DICER1 mutations.
- Central nervous system (CNS) neuroblastoma with FOXR2 activation (CNS NB-FOXR2): Representing 10% to 15% of nonmedulloblastoma embryonal tumors, this tumor may occur in children younger than 3 years, but it more frequently occurs in older children. This subtype is characterized by genomic alterations that lead to increased expression of the transcription factor FOXR2. CNS NB-FOXR2 is primarily observed in children younger than 10 years, and the histology of these tumors is typically that of CNS neuroblastoma or CNS ganglioneuroblastoma. There is no single genomic alteration among CNS NB-FOXR2 tumors leading to FOXR2 overexpression, with gene fusions involving multiple FOXR2 partners identified. Protein expression of SOX10 and ANKRD55 detected by immunohistochemistry has been proposed as a potential biomarker to differentiate CNS NB-FOXR2 tumors from related tumor types.
- CNS high-grade neuroepithelial tumor with BCOR alteration (CNS HGNET-BCOR): Representing up to 3% of nonmedulloblastoma embryonal tumors, this subtype is characterized by internal tandem duplications of BCOR, a genomic alteration that is also found in clear cell sarcoma of the kidney. While the median age at diagnosis is younger than 10 years, cases arising in the second decade of life and beyond do occur.
Although not listed as separate entities in the 2021 WHO Classification of Tumours of the CNS, other nonmedulloblastoma embryonal tumors have also been described as separate entities, including the following:
- CNS Ewing sarcoma family tumor with CIC alteration (CNS EFT-CIC): Representing 2% to 4% of nonmedulloblastoma embryonal tumors, this subtype is characterized by genomic alterations affecting CIC (located on chromosome 19q13.2), with fusion to NUTM1 being identified in several cases tested. CIC gene fusions are also identified in extra-CNS Ewing-like sarcomas, and the gene expression signature of CNS EFT-CIC tumors is similar to that of these sarcomas. CNS EFT-CIC tumors generally occur in children younger than 10 years and are characterized by a small cell phenotype but with variable histology.
- CNS high-grade neuroepithelial tumor with MN1 alteration (CNS HGNET-MN1): Representing 1% to 3% of nonmedulloblastoma embryonal tumors, this subtype is characterized by gene fusions involving MN1 (located on chromosome 22q12.3), with fusion partners including BEND2 and CXXC5. The CNS HGNET-MN1 subtype shows a striking female predominance, and it tends to occur in the second decade of life. This subtype contained most cases carrying a diagnosis of astroblastoma per the 2007 WHO classification scheme. This subtype has not been added to the WHO diagnostic lexicon. Two other reports that together examined 35 cases of histologically defined astroblastoma found that 14 showed methylation profiles consistent with CNS HGNET-MN1 and/or MN1 alterations by fluorescence in situ hybridization.
- Medulloepithelioma: Medulloepithelioma with the classic C19MC amplification is considered an ETMR, C19MC-altered (see the ETMR information above). However, when a tumor has the histological features of medulloepithelioma, but without a C19MC amplification, it is still identified as an ETMR. Medulloepithelioma tumors are rare and tend to arise most commonly in infants and young children. Medulloepitheliomas, which histologically recapitulate the embryonal neural tube, tend to arise supratentorially, primarily intraventricularly, but may arise infratentorially, in the cauda, and even extraneurally, along nerve roots. Intraocular medulloepithelioma is biologically distinct from intra-axial medulloepithelioma.
- CNS embryonal tumor with PLAGL amplification: A retrospective analysis of more than 90,000 pediatric and adult brain tumors identified a small subset of embryonal tumors (n = 31) with distinct methylation profiles and high-level amplification and overexpression of either PLAGL1 or PLAGL2. Additional recurrent genetic alterations observed in other pediatric CNS tumor types were not observed in these cases. These tumors occurred throughout the brain and were most commonly composed of primitive embryonal-like cells without markers of glial or neuronal differentiation. In this small cohort, differences in age at diagnosis and 10-year overall survival (OS) were reported between patients with PLAGL1 amplification (median age, 10.5 years; OS rate, 66%), compared with PLAGL2 amplification (median age, 2 years; OS rate, 25%).
For information about the treatment of childhood PNETs, see Childhood Medulloblastoma and Other Central Nervous System Embryonal Tumors Treatment.
Pineoblastoma
Genomics of Pineoblastoma
Pineoblastoma, which was previously conventionally grouped with embryonal tumors, is now categorized by the World Health Organization (WHO) as a pineal parenchymal tumor. Given that therapies for pineoblastoma are quite similar to those used for embryonal tumors, the previous convention of including pineoblastoma with the central nervous system (CNS) embryonal tumors is followed here. Pineoblastoma is associated with germline mutations in both the RB1 gene and in DICER1, as described below:
- Pineoblastoma is associated with germline mutations in RB1, with the term trilateral retinoblastoma used to refer to ocular retinoblastoma in combination with a histologically similar brain tumor generally arising in the pineal gland or other midline structures. Historically, intracranial tumors have been reported in 5% to 15% of children with heritable retinoblastoma. Rates of pineoblastoma among children with heritable retinoblastoma who undergo current treatment programs may be lower than these historical estimates.
- Germline DICER1 mutations have also been reported in patients with pineoblastoma. Among 18 patients with pineoblastoma, 3 patients with DICER1 germline mutations were identified, and an additional 3 patients known to be carriers of germline DICER1 mutations developed pineoblastoma. The DICER1 mutations in patients with pineoblastoma are loss-of-function mutations that appear to be distinct from the mutations observed in DICER1 syndrome–related tumors such as pleuropulmonary blastoma.
Genomic methods have been applied to pineoblastoma in an attempt to learn more about the tumor biology and guide future molecular classification. A retrospective, international, meta-analysis included 221 children and adults diagnosed with pineoblastoma and pineal parenchymal tumors of intermediate differentiation. The evaluation identified four molecular groups of pineoblastoma based on DNA methylation and transcriptome profiling. These groups were termed PB-miRNA1 and PB-miRNA2 (with recurrent alterations in microRNA processing genes), PB-MYC/FOXR2 (with MYC amplification and FOXR2 overexpression) and PB-RB1 (with RB1 alterations). A fifth distinct group of tumors (comprised of both histological pineoblastomas and pineal parenchymal tumors of intermediate differentiation) had recurrent KBTBD4 mutations and was designated pineal parenchymal tumors of intermediate differentiation. Further studies will be necessary to refine these molecular groups and their clinical implications.
For information about the treatment of childhood pineoblastoma, see Childhood Medulloblastoma and Other Central Nervous System Embryonal Tumors Treatment.
Ependymomas
Molecular Subgroups of Ependymoma
Molecular characterization studies have previously identified nine molecular subgroups of ependymoma, six of which predominate in childhood. The subgroups are determined by their distinctive DNA methylation and gene expression profiles and unique spectrum of genomic alterations (see Figure 6).
One new molecularly defined ependymoma was added to the 2021 World Health Organization (WHO) Classification of Tumours of the Central Nervous System: spinal ependymoma with MYCN amplification. The 2021 classification further described ependymal tumors defined by anatomical location and histology but not by molecular alteration. These tumors are called posterior fossa ependymoma (PF-EPN), supratentorial ependymoma (ST-EPN), and spinal ependymoma (SP-EPN). These tumors either contain a unique molecular alteration (not elsewhere classified [NEC]) or their molecular analysis failed or was not obtained (not otherwise specified [NOS]).
- Infratentorial tumors.
- Posterior fossa ependymoma (PF-EPN).
- Posterior fossa A (PF-EPN-A), loss of H3 K27 trimethylation mark.
- Posterior fossa B (PF-EPN-B), retained H3 K27 trimethylation mark.
- Supratentorial tumors.
- Supratentorial ependymoma (ST-EPN).
- ZFTA fusion–positive ependymoma (ST-EPN-ZFTA). This was previously called RELA fusion–positive ependymoma (ST-EPN-RELA), but it was renamed because ZFTA is the new designation for C11orf95, and ZFTA may be fused with a partner gene other than RELA.
- YAP1 fusion–positive ependymoma (ST-EPN-YAP1).
- Spinal tumors.
- Spinal ependymoma (SP-EPN).
- Spinal ependymoma, MYCN-amplified (SP-EPN-MYCN).
- Myxopapillary ependymoma (SP-EPN-MPE).
Subependymoma—whether supratentorial, infratentorial, or spinal—accounts for the remaining three molecular variants, and it is rarely, if ever, seen in children.
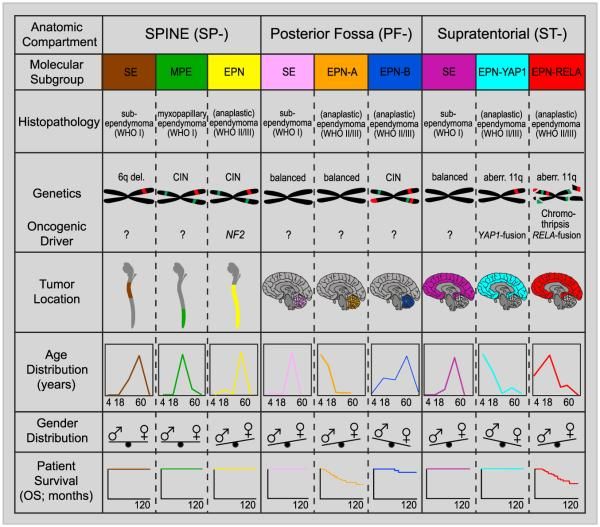 Figure 6. Graphical summary of key molecular and clinical characteristics of ependymal tumor subgroups. Schematic representation of key genetic and epigenetic findings in the nine molecular subgroups of ependymal tumors as identified by methylation profiling. CIN, Chromosomal instability. Reprinted from Cancer Cell, Volume 27, Kristian W. Pajtler, Hendrik Witt, Martin Sill, David T.W. Jones, Volker Hovestadt, Fabian Kratochwil,
Khalida Wani, Ruth Tatevossian, Chandanamali Punchihewa, Pascal Johann, Juri Reimand, Hans-Jorg Warnatz,
Marina Ryzhova, Steve Mack, Vijay Ramaswamy, David Capper, Leonille Schweizer, Laura Sieber,
Andrea Wittmann, Zhiqin Huang, Peter van Sluis, Richard Volckmann, Jan Koster, Rogier Versteeg,
Daniel Fults, Helen Toledano, Smadar Avigad, Lindsey M. Hoffman, Andrew M. Donson, Nicholas Foreman,
Ekkehard Hewer, Karel Zitterbart, Mark Gilbert, Terri S. Armstrong, Nalin Gupta, Jeffrey C. Allen,
Matthias A. Karajannis, David Zagzag, Martin Hasselblatt, Andreas E. Kulozik, Olaf Witt, V. Peter Collins,
Katja von Hoff, Stefan Rutkowski, Torsten Pietsch, Gary Bader, Marie-Laure Yaspo, Andreas von Deimling,
Peter Lichter, Michael D. Taylor, Richard Gilbertson, David W. Ellison, Kenneth Aldape, Andrey Korshunov,
Marcel Kool, and Stefan M. Pfister, Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups, Pages 728–743, Copyright (2015), with permission from Elsevier.
Figure 6. Graphical summary of key molecular and clinical characteristics of ependymal tumor subgroups. Schematic representation of key genetic and epigenetic findings in the nine molecular subgroups of ependymal tumors as identified by methylation profiling. CIN, Chromosomal instability. Reprinted from Cancer Cell, Volume 27, Kristian W. Pajtler, Hendrik Witt, Martin Sill, David T.W. Jones, Volker Hovestadt, Fabian Kratochwil,
Khalida Wani, Ruth Tatevossian, Chandanamali Punchihewa, Pascal Johann, Juri Reimand, Hans-Jorg Warnatz,
Marina Ryzhova, Steve Mack, Vijay Ramaswamy, David Capper, Leonille Schweizer, Laura Sieber,
Andrea Wittmann, Zhiqin Huang, Peter van Sluis, Richard Volckmann, Jan Koster, Rogier Versteeg,
Daniel Fults, Helen Toledano, Smadar Avigad, Lindsey M. Hoffman, Andrew M. Donson, Nicholas Foreman,
Ekkehard Hewer, Karel Zitterbart, Mark Gilbert, Terri S. Armstrong, Nalin Gupta, Jeffrey C. Allen,
Matthias A. Karajannis, David Zagzag, Martin Hasselblatt, Andreas E. Kulozik, Olaf Witt, V. Peter Collins,
Katja von Hoff, Stefan Rutkowski, Torsten Pietsch, Gary Bader, Marie-Laure Yaspo, Andreas von Deimling,
Peter Lichter, Michael D. Taylor, Richard Gilbertson, David W. Ellison, Kenneth Aldape, Andrey Korshunov,
Marcel Kool, and Stefan M. Pfister, Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups, Pages 728–743, Copyright (2015), with permission from Elsevier.
Infratentorial tumors
Posterior fossa A ependymoma (PF-EPN-A)
The most common posterior fossa ependymoma subgroup is PF-EPN-A and is characterized by the following:
- Presentation in young children (median age, 3 years).
- Low rates of variants that affect protein structure, approximately five per genome.
- Gain of chromosome 1q, a known poor prognostic factor for patients with ependymoma, in approximately 25% of cases.
- Loss of chromosome 6q, reported to be a poor prognostic factor for patients with PF-EPN-A, in 8% to 10% of cases.
- A balanced chromosomal profile with few chromosomal gains or losses.
- Loss of the H3 K27 trimethylation mark and globally hypomethylated DNA. A prospective multi-institutional study analyzed 147 patients with ependymoma. The study reported high sensitivity and specificity for immunohistochemical detection of loss of the H3 K27 trimethylation mark in identifying PF-EPN-A cases. Loss of this mark occurs through multiple mechanisms, including the following:
- Recurrent variants of EZHIP in 10% of cases, with high EZHIP mRNA expression across almost all PF-EPN-A. EZHIP expression (with or without alteration) results in inhibition of the methyltransferase EZH2 leading to loss of the H3 K27 trimethylation mark.
- Recurrent K27M variants in histone H3 variants in a small proportion of cases. Unlike diffuse midline gliomas, variants in H3.1 (H3C2 and H3C3) are more common than variants in H3.3 (H3-3A). Histone variants are mutually exclusive with high expression of EZHIP, and they also lead to loss of the H3 K27 trimethylation mark through EZH2 inhibition.
A study that included over 600 cases of PF-EPN-A used methylation array profiling to divide this population into two distinctive subgroups, PFA-1 and PFA-2. Gene expression profiling suggested that these two subtypes may arise in different anatomical locations in the hindbrain. Within both PFA-1 and PFA-2 groups, distinctive minor subtypes could be identified, suggesting the presence of heterogeneity. Additional study will be required to define the clinical significance of these subtypes.
Posterior fossa B ependymoma (PF-EPN-B)
The PF-EPN-B subgroup is less common than the PF-EPN-A subgroup, representing 15% to 20% of all posterior fossa ependymomas in children. PF-EPN-B is characterized by the following:
- Presentation primarily in adolescents and young adults (median age, 30 years).
- Low rates of variants that affect protein structure (approximately five per genome), with no recurring variants.
- Numerous cytogenetic abnormalities, primarily involving the gain/loss of whole chromosomes.
- Retained H3 K27 trimethylation.
- 1q gain and 6q loss occur in PF-EPN-B but have not been reported as prognostic in this subgroup (unlike in PF-EPN-A).
Supratentorial tumors
Supratentorial ependymomas with ZFTA fusions (ST-EPN-ZFTA)
ST-EPN-ZFTA is the largest subset of pediatric supratentorial ependymomas and is predominantly characterized by gene fusions involving RELA, a transcriptional factor important in NF-κB pathway activity. ST-EPN-ZFTA is characterized by the following:
- Represents approximately 70% of supratentorial ependymomas in children, and presents at a median age of 8 years.
- Presence of ZFTA fusions result from chromothripsis involving chromosome 11q13.1.
- Low rates of variants that affect protein structure and near absence of recurring variants outside of ZFTA::RELA fusions.
- Evidence of NF-κB pathway activation at the protein and RNA level.
- Gain of chromosome 1q, in approximately one-quarter of cases, with an indeterminate effect on survival.
- The concordance was high between immunohistochemistry for nuclear p65-RelA, fluorescence in situ hybridization for ZFTA and RELA, and DNA methylation-based classification for defining ST-EPN-ZFTA.
- Homozygous deletion of CDKN2A has been associated with a poor prognosis in patients with ZFTA fusion–positive ependymoma.[Level of evidence B4] CDKN2A deletion has also been reported as a secondary event in recurrent ependymoma.
Supratentorial ependymomas with YAP1 fusions (ST-EPN-YAP1)
ST-EPN-YAP1 is the second, less common subset of supratentorial ependymomas and has fusions involving YAP1 on chromosome 11. ST-EPN-YAP1 is characterized by the following:
- Median age at diagnosis of 1.4 years.
- Presence of a gene fusion involving YAP1, with MAMLD1 being the most common fusion partner.
- A relatively stable genome with few chromosomal changes other than the YAP1 fusion.
Supratentorial ependymomas without ZFTA or YAP1 fusions (on chromosome 11) are an undefined entity, and it is unclear what these samples represent. By DNA methylation analysis, these samples often cluster with other entities such as high-grade gliomas and embryonal tumors. Caution should be taken when diagnosing a supratentorial ependymoma that does not harbor a fusion involving chromosome 11.
Spinal ependymoma with MYCN amplification (SP-EPN-MYCN)
SP-EPN-MYCN is rare, with only 27 cases reported.
- Median age at presentation was 31 years (range, 12–56 years).
- High level of MYCN amplification was present at diagnosis and relapse.
- SP-EPN-MYCN has a unique methylation profile compared with other spinal cord ependymomas, MYCN-amplified pediatric-type glioblastoma, and neuroblastoma.
For information about the treatment of childhood ependymoma, see Childhood Ependymoma Treatment.
References
- Louis DN, Perry A, Wesseling P, et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23 (8): 1231-1251, 2021.
- Zhang J, Wu G, Miller CP, et al.: Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45 (6): 602-12, 2013.
- Ramkissoon LA, Horowitz PM, Craig JM, et al.: Genomic analysis of diffuse pediatric low-grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proc Natl Acad Sci U S A 110 (20): 8188-93, 2013.
- Qaddoumi I, Orisme W, Wen J, et al.: Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol 131 (6): 833-45, 2016.
- Pollack IF, Hamilton RL, Sobol RW, et al.: IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children's Oncology Group. Childs Nerv Syst 27 (1): 87-94, 2011.
- Packer RJ, Iavarone A, Jones DTW, et al.: Implications of new understandings of gliomas in children and adults with NF1: report of a consensus conference. Neuro Oncol 22 (6): 773-784, 2020.
- Ryall S, Zapotocky M, Fukuoka K, et al.: Integrated Molecular and Clinical Analysis of 1,000 Pediatric Low-Grade Gliomas. Cancer Cell 37 (4): 569-583.e5, 2020.
- D'Angelo F, Ceccarelli M, Tala, et al.: The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat Med 25 (1): 176-187, 2019.
- Franz DN, Agricola K, Mays M, et al.: Everolimus for subependymal giant cell astrocytoma: 5-year final analysis. Ann Neurol 78 (6): 929-38, 2015.
- Mackay A, Burford A, Carvalho D, et al.: Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 32 (4): 520-537.e5, 2017.
- Bouffet E, Larouche V, Campbell BB, et al.: Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol 34 (19): 2206-11, 2016.
- Das A, Tabori U, Sambira Nahum LC, et al.: Efficacy of Nivolumab in Pediatric Cancers with High Mutation Burden and Mismatch Repair Deficiency. Clin Cancer Res 29 (23): 4770-4783, 2023.
- Jones DT, Kocialkowski S, Liu L, et al.: Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68 (21): 8673-7, 2008.
- Hawkins C, Walker E, Mohamed N, et al.: BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res 17 (14): 4790-8, 2011.
- Mistry M, Zhukova N, Merico D, et al.: BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol 33 (9): 1015-22, 2015.
- López GY, Van Ziffle J, Onodera C, et al.: The genetic landscape of gliomas arising after therapeutic radiation. Acta Neuropathol 137 (1): 139-150, 2019.
- Lassaletta A, Zapotocky M, Mistry M, et al.: Therapeutic and Prognostic Implications of BRAF V600E in Pediatric Low-Grade Gliomas. J Clin Oncol 35 (25): 2934-2941, 2017.
- Ho CY, Mobley BC, Gordish-Dressman H, et al.: A clinicopathologic study of diencephalic pediatric low-grade gliomas with BRAF V600 mutation. Acta Neuropathol 130 (4): 575-85, 2015.
- Guerreiro Stucklin AS, Ryall S, Fukuoka K, et al.: Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun 10 (1): 4343, 2019.
- Clarke M, Mackay A, Ismer B, et al.: Infant High-Grade Gliomas Comprise Multiple Subgroups Characterized by Novel Targetable Gene Fusions and Favorable Outcomes. Cancer Discov 10 (7): 942-963, 2020.
- Meredith DM, Cooley LD, Dubuc A, et al.: ROS1 Alterations as a Potential Driver of Gliomas in Infant, Pediatric, and Adult Patients. Mod Pathol 36 (11): 100294, 2023.
- Jones DT, Hutter B, Jäger N, et al.: Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45 (8): 927-32, 2013.
- Louis DN, Perry A, Reifenberger G, et al.: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016.
- Bandopadhayay P, Ramkissoon LA, Jain P, et al.: MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet 48 (3): 273-82, 2016.
- D'Aronco L, Rouleau C, Gayden T, et al.: Brainstem angiocentric gliomas with MYB-QKI rearrangements. Acta Neuropathol 134 (4): 667-669, 2017.
- Chan E, Bollen AW, Sirohi D, et al.: Angiocentric glioma with MYB-QKI fusion located in the brainstem, rather than cerebral cortex. Acta Neuropathol 134 (4): 671-673, 2017.
- Sturm D, Orr BA, Toprak UH, et al.: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5): 1060-72, 2016.
- Lehman NL, Usubalieva A, Lin T, et al.: Genomic analysis demonstrates that histologically-defined astroblastomas are molecularly heterogeneous and that tumors with MN1 rearrangement exhibit the most favorable prognosis. Acta Neuropathol Commun 7 (1): 42, 2019.
- Wood MD, Tihan T, Perry A, et al.: Multimodal molecular analysis of astroblastoma enables reclassification of most cases into more specific molecular entities. Brain Pathol 28 (2): 192-202, 2018.
- Hirose T, Nobusawa S, Sugiyama K, et al.: Astroblastoma: a distinct tumor entity characterized by alterations of the X chromosome and MN1 rearrangement. Brain Pathol 28 (5): 684-694, 2018.
- Lucas CG, Solomon DA, Perry A: A review of recently described genetic alterations in central nervous system tumors. Hum Pathol 96: 56-66, 2020.
- Paugh BS, Qu C, Jones C, et al.: Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol 28 (18): 3061-8, 2010.
- Bax DA, Mackay A, Little SE, et al.: A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin Cancer Res 16 (13): 3368-77, 2010.
- Ward SJ, Karakoula K, Phipps KP, et al.: Cytogenetic analysis of paediatric astrocytoma using comparative genomic hybridisation and fluorescence in-situ hybridisation. J Neurooncol 98 (3): 305-18, 2010.
- Sturm D, Witt H, Hovestadt V, et al.: Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22 (4): 425-37, 2012.
- Korshunov A, Ryzhova M, Hovestadt V, et al.: Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol 129 (5): 669-78, 2015.
- Castel D, Kergrohen T, Tauziède-Espariat A, et al.: Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol 139 (6): 1109-1113, 2020.
- Rodriguez Gutierrez D, Jones C, Varlet P, et al.: Radiological Evaluation of Newly Diagnosed Non-Brainstem Pediatric High-Grade Glioma in the HERBY Phase II Trial. Clin Cancer Res 26 (8): 1856-1865, 2020.
- Buczkowicz P, Hoeman C, Rakopoulos P, et al.: Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46 (5): 451-6, 2014.
- Taylor KR, Mackay A, Truffaux N, et al.: Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46 (5): 457-61, 2014.
- Auffret L, Ajlil Y, Tauziède-Espariat A, et al.: A new subtype of diffuse midline glioma, H3 K27 and BRAF/FGFR1 co-altered: a clinico-radiological and histomolecular characterisation. Acta Neuropathol 147 (1): 2, 2023.
- Williams EA, Brastianos PK, Wakimoto H, et al.: A comprehensive genomic study of 390 H3F3A-mutant pediatric and adult diffuse high-grade gliomas, CNS WHO grade 4. Acta Neuropathol 146 (3): 515-525, 2023.
- Jain SU, Do TJ, Lund PJ, et al.: PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat Commun 10 (1): 2146, 2019.
- Hübner JM, Müller T, Papageorgiou DN, et al.: EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol 21 (7): 878-889, 2019.
- Chen CCL, Deshmukh S, Jessa S, et al.: Histone H3.3G34-Mutant Interneuron Progenitors Co-opt PDGFRA for Gliomagenesis. Cell 183 (6): 1617-1633.e22, 2020.
- Mackay A, Burford A, Molinari V, et al.: Molecular, Pathological, Radiological, and Immune Profiling of Non-brainstem Pediatric High-Grade Glioma from the HERBY Phase II Randomized Trial. Cancer Cell 33 (5): 829-842.e5, 2018.
- Yeo KK, Alexandrescu S, Cotter JA, et al.: Multi-institutional study of the frequency, genomic landscape, and outcome of IDH-mutant glioma in pediatrics. Neuro Oncol 25 (1): 199-210, 2023.
- Suwala AK, Stichel D, Schrimpf D, et al.: Primary mismatch repair deficient IDH-mutant astrocytoma (PMMRDIA) is a distinct type with a poor prognosis. Acta Neuropathol 141 (1): 85-100, 2021.
- Reinhardt A, Stichel D, Schrimpf D, et al.: Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol 136 (2): 273-291, 2018.
- Korshunov A, Schrimpf D, Ryzhova M, et al.: H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol 134 (3): 507-516, 2017.
- Becker AJ: Ganglioglioma. In: Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. IARC Press, 2016, pp 138-41.
- Blumcke I, Spreafico R, Haaker G, et al.: Histopathological Findings in Brain Tissue Obtained during Epilepsy Surgery. N Engl J Med 377 (17): 1648-1656, 2017.
- Pekmezci M, Villanueva-Meyer JE, Goode B, et al.: The genetic landscape of ganglioglioma. Acta Neuropathol Commun 6 (1): 47, 2018.
- Bianchi F, Tamburrini G, Massimi L, et al.: Supratentorial tumors typical of the infantile age: desmoplastic infantile ganglioglioma (DIG) and astrocytoma (DIA). A review. Childs Nerv Syst 32 (10): 1833-8, 2016.
- Trehan G, Bruge H, Vinchon M, et al.: MR imaging in the diagnosis of desmoplastic infantile tumor: retrospective study of six cases. AJNR Am J Neuroradiol 25 (6): 1028-33, 2004 Jun-Jul.
- Wang AC, Jones DTW, Abecassis IJ, et al.: Desmoplastic Infantile Ganglioglioma/Astrocytoma (DIG/DIA) Are Distinct Entities with Frequent BRAFV600 Mutations. Mol Cancer Res 16 (10): 1491-1498, 2018.
- Blessing MM, Blackburn PR, Krishnan C, et al.: Desmoplastic Infantile Ganglioglioma: A MAPK Pathway-Driven and Microglia/Macrophage-Rich Neuroepithelial Tumor. J Neuropathol Exp Neurol 78 (11): 1011-1021, 2019.
- Greer A, Foreman NK, Donson A, et al.: Desmoplastic infantile astrocytoma/ganglioglioma with rare BRAF V600D mutation. Pediatr Blood Cancer 64 (6): , 2017.
- Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. IARC Press, 2016.
- Stone TJ, Keeley A, Virasami A, et al.: Comprehensive molecular characterisation of epilepsy-associated glioneuronal tumours. Acta Neuropathol 135 (1): 115-129, 2018.
- Rivera B, Gayden T, Carrot-Zhang J, et al.: Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors. Acta Neuropathol 131 (6): 847-63, 2016.
- Matsumura N, Nobusawa S, Ito J, et al.: Multiplex ligation-dependent probe amplification analysis is useful for detecting a copy number gain of the FGFR1 tyrosine kinase domain in dysembryoplastic neuroepithelial tumors. J Neurooncol 143 (1): 27-33, 2019.
- Pages M, Lacroix L, Tauziede-Espariat A, et al.: Papillary glioneuronal tumors: histological and molecular characteristics and diagnostic value of SLC44A1-PRKCA fusion. Acta Neuropathol Commun 3: 85, 2015.
- Bridge JA, Liu XQ, Sumegi J, et al.: Identification of a novel, recurrent SLC44A1-PRKCA fusion in papillary glioneuronal tumor. Brain Pathol 23 (2): 121-8, 2013.
- Hou Y, Pinheiro J, Sahm F, et al.: Papillary glioneuronal tumor (PGNT) exhibits a characteristic methylation profile and fusions involving PRKCA. Acta Neuropathol 137 (5): 837-846, 2019.
- Sievers P, Appay R, Schrimpf D, et al.: Rosette-forming glioneuronal tumors share a distinct DNA methylation profile and mutations in FGFR1, with recurrent co-mutation of PIK3CA and NF1. Acta Neuropathol 138 (3): 497-504, 2019.
- Deng MY, Sill M, Chiang J, et al.: Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol 136 (2): 239-253, 2018.
- Chiang JCH, Harreld JH, Orr BA, et al.: Low-grade spinal glioneuronal tumors with BRAF gene fusion and 1p deletion but without leptomeningeal dissemination. Acta Neuropathol 134 (1): 159-162, 2017.
- Chiang J, Dalton J, Upadhyaya SA, et al.: Chromosome arm 1q gain is an adverse prognostic factor in localized and diffuse leptomeningeal glioneuronal tumors with BRAF gene fusion and 1p deletion. Acta Neuropathol 137 (1): 179-181, 2019.
- Sievers P, Stichel D, Schrimpf D, et al.: FGFR1:TACC1 fusion is a frequent event in molecularly defined extraventricular neurocytoma. Acta Neuropathol 136 (2): 293-302, 2018.
- Biegel JA, Tan L, Zhang F, et al.: Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res 8 (11): 3461-7, 2002.
- Biegel JA, Kalpana G, Knudsen ES, et al.: The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer Res 62 (1): 323-8, 2002.
- Schneppenheim R, Frühwald MC, Gesk S, et al.: Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet 86 (2): 279-84, 2010.
- Hasselblatt M, Gesk S, Oyen F, et al.: Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 35 (6): 933-5, 2011.
- Hasselblatt M, Nagel I, Oyen F, et al.: SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol 128 (3): 453-6, 2014.
- WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
- Lee RS, Stewart C, Carter SL, et al.: A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122 (8): 2983-8, 2012.
- Kieran MW, Roberts CW, Chi SN, et al.: Absence of oncogenic canonical pathway mutations in aggressive pediatric rhabdoid tumors. Pediatr Blood Cancer 59 (7): 1155-7, 2012.
- Hasselblatt M, Isken S, Linge A, et al.: High-resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors. Genes Chromosomes Cancer 52 (2): 185-90, 2013.
- Torchia J, Picard D, Lafay-Cousin L, et al.: Molecular subgroups of atypical teratoid rhabdoid tumours in children: an integrated genomic and clinicopathological analysis. Lancet Oncol 16 (5): 569-82, 2015.
- Johann PD, Erkek S, Zapatka M, et al.: Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 29 (3): 379-93, 2016.
- Upadhyaya SA, Robinson GW, Onar-Thomas A, et al.: Relevance of Molecular Groups in Children with Newly Diagnosed Atypical Teratoid Rhabdoid Tumor: Results from Prospective St. Jude Multi-institutional Trials. Clin Cancer Res 27 (10): 2879-2889, 2021.
- Johann PD, Hovestadt V, Thomas C, et al.: Cribriform neuroepithelial tumor: molecular characterization of a SMARCB1-deficient non-rhabdoid tumor with favorable long-term outcome. Brain Pathol 27 (4): 411-418, 2017.
- Frühwald MC, Hasselblatt M, Nemes K, et al.: Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro Oncol 22 (7): 1006-1017, 2020.
- Federico A, Thomas C, Miskiewicz K, et al.: ATRT-SHH comprises three molecular subgroups with characteristic clinical and histopathological features and prognostic significance. Acta Neuropathol 143 (6): 697-711, 2022.
- Wilson BG, Wang X, Shen X, et al.: Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18 (4): 316-28, 2010.
- Knutson SK, Warholic NM, Wigle TJ, et al.: Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110 (19): 7922-7, 2013.
- Kurmasheva RT, Sammons M, Favours E, et al.: Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 64 (3): , 2017.
- Italiano A, Soria JC, Toulmonde M, et al.: Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 19 (5): 649-659, 2018.
- Onvani S, Etame AB, Smith CA, et al.: Genetics of medulloblastoma: clues for novel therapies. Expert Rev Neurother 10 (5): 811-23, 2010.
- Dubuc AM, Northcott PA, Mack S, et al.: The genetics of pediatric brain tumors. Curr Neurol Neurosci Rep 10 (3): 215-23, 2010.
- Thompson MC, Fuller C, Hogg TL, et al.: Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24 (12): 1924-31, 2006.
- Kool M, Koster J, Bunt J, et al.: Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One 3 (8): e3088, 2008.
- Tabori U, Baskin B, Shago M, et al.: Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol 28 (8): 1345-50, 2010.
- Pfister S, Remke M, Benner A, et al.: Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol 27 (10): 1627-36, 2009.
- Ellison DW, Onilude OE, Lindsey JC, et al.: beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children's Cancer Study Group Brain Tumour Committee. J Clin Oncol 23 (31): 7951-7, 2005.
- Polkinghorn WR, Tarbell NJ: Medulloblastoma: tumorigenesis, current clinical paradigm, and efforts to improve risk stratification. Nat Clin Pract Oncol 4 (5): 295-304, 2007.
- Giangaspero F, Wellek S, Masuoka J, et al.: Stratification of medulloblastoma on the basis of histopathological grading. Acta Neuropathol 112 (1): 5-12, 2006.
- Northcott PA, Korshunov A, Witt H, et al.: Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29 (11): 1408-14, 2011.
- Pomeroy SL, Tamayo P, Gaasenbeek M, et al.: Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 415 (6870): 436-42, 2002.
- Jones DT, Jäger N, Kool M, et al.: Dissecting the genomic complexity underlying medulloblastoma. Nature 488 (7409): 100-5, 2012.
- Peyrl A, Chocholous M, Kieran MW, et al.: Antiangiogenic metronomic therapy for children with recurrent embryonal brain tumors. Pediatr Blood Cancer 59 (3): 511-7, 2012.
- Taylor MD, Northcott PA, Korshunov A, et al.: Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123 (4): 465-72, 2012.
- Kool M, Korshunov A, Remke M, et al.: Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 123 (4): 473-84, 2012.
- Pietsch T, Schmidt R, Remke M, et al.: Prognostic significance of clinical, histopathological, and molecular characteristics of medulloblastomas in the prospective HIT2000 multicenter clinical trial cohort. Acta Neuropathol 128 (1): 137-49, 2014.
- Cho YJ, Tsherniak A, Tamayo P, et al.: Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 29 (11): 1424-30, 2011.
- Gajjar A, Bowers DC, Karajannis MA, et al.: Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape. J Clin Oncol 33 (27): 2986-98, 2015.
- Morrissy AS, Cavalli FMG, Remke M, et al.: Spatial heterogeneity in medulloblastoma. Nat Genet 49 (5): 780-788, 2017.
- Wang X, Dubuc AM, Ramaswamy V, et al.: Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol 129 (3): 449-57, 2015.
- Cavalli FMG, Remke M, Rampasek L, et al.: Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 31 (6): 737-754.e6, 2017.
- Northcott PA, Buchhalter I, Morrissy AS, et al.: The whole-genome landscape of medulloblastoma subtypes. Nature 547 (7663): 311-317, 2017.
- Schwalbe EC, Lindsey JC, Nakjang S, et al.: Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol 18 (7): 958-971, 2017.
- Hicks D, Rafiee G, Schwalbe EC, et al.: The molecular landscape and associated clinical experience in infant medulloblastoma: prognostic significance of second-generation subtypes. Neuropathol Appl Neurobiol 47 (2): 236-250, 2021.
- Northcott PA, Jones DT, Kool M, et al.: Medulloblastomics: the end of the beginning. Nat Rev Cancer 12 (12): 818-34, 2012.
- Korshunov A, Sahm F, Zheludkova O, et al.: DNA methylation profiling is a method of choice for molecular verification of pediatric WNT-activated medulloblastomas. Neuro Oncol 21 (2): 214-221, 2019.
- Gibson P, Tong Y, Robinson G, et al.: Subtypes of medulloblastoma have distinct developmental origins. Nature 468 (7327): 1095-9, 2010.
- Ellison DW, Dalton J, Kocak M, et al.: Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol 121 (3): 381-96, 2011.
- Gajjar A, Chintagumpala M, Ashley D, et al.: Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol 7 (10): 813-20, 2006.
- Michalski JM, Janss AJ, Vezina LG, et al.: Children's Oncology Group Phase III Trial of Reduced-Dose and Reduced-Volume Radiotherapy With Chemotherapy for Newly Diagnosed Average-Risk Medulloblastoma. J Clin Oncol 39 (24): 2685-2697, 2021.
- Goschzik T, Mynarek M, Doerner E, et al.: Genetic alterations of TP53 and OTX2 indicate increased risk of relapse in WNT medulloblastomas. Acta Neuropathol 144 (6): 1143-1156, 2022.
- Begemann M, Waszak SM, Robinson GW, et al.: Germline GPR161 Mutations Predispose to Pediatric Medulloblastoma. J Clin Oncol 38 (1): 43-50, 2020.
- Shuai S, Suzuki H, Diaz-Navarro A, et al.: The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 574 (7780): 712-716, 2019.
- Suzuki H, Kumar SA, Shuai S, et al.: Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature 574 (7780): 707-711, 2019.
- Kool M, Jones DT, Jäger N, et al.: Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 25 (3): 393-405, 2014.
- Robinson GW, Rudneva VA, Buchhalter I, et al.: Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol 19 (6): 768-784, 2018.
- Lafay-Cousin L, Bouffet E, Strother D, et al.: Phase II Study of Nonmetastatic Desmoplastic Medulloblastoma in Children Younger Than 4 Years of Age: A Report of the Children's Oncology Group (ACNS1221). J Clin Oncol 38 (3): 223-231, 2020.
- Leary SE, Zhou T, Holmes E, et al.: Histology predicts a favorable outcome in young children with desmoplastic medulloblastoma: a report from the children's oncology group. Cancer 117 (14): 3262-7, 2011.
- Giangaspero F, Perilongo G, Fondelli MP, et al.: Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg 91 (6): 971-7, 1999.
- Rutkowski S, von Hoff K, Emser A, et al.: Survival and prognostic factors of early childhood medulloblastoma: an international meta-analysis. J Clin Oncol 28 (33): 4961-8, 2010.
- Garrè ML, Cama A, Bagnasco F, et al.: Medulloblastoma variants: age-dependent occurrence and relation to Gorlin syndrome--a new clinical perspective. Clin Cancer Res 15 (7): 2463-71, 2009.
- von Bueren AO, von Hoff K, Pietsch T, et al.: Treatment of young children with localized medulloblastoma by chemotherapy alone: results of the prospective, multicenter trial HIT 2000 confirming the prognostic impact of histology. Neuro Oncol 13 (6): 669-79, 2011.
- Shih DJ, Northcott PA, Remke M, et al.: Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol 32 (9): 886-96, 2014.
- Schwalbe EC, Williamson D, Lindsey JC, et al.: DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin-fixed biopsies. Acta Neuropathol 125 (3): 359-71, 2013.
- Zhukova N, Ramaswamy V, Remke M, et al.: Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol 31 (23): 2927-35, 2013.
- Gajjar A, Robinson GW, Smith KS, et al.: Outcomes by Clinical and Molecular Features in Children With Medulloblastoma Treated With Risk-Adapted Therapy: Results of an International Phase III Trial (SJMB03). J Clin Oncol 39 (7): 822-835, 2021.
- Goschzik T, Schwalbe EC, Hicks D, et al.: Prognostic effect of whole chromosomal aberration signatures in standard-risk, non-WNT/non-SHH medulloblastoma: a retrospective, molecular analysis of the HIT-SIOP PNET 4 trial. Lancet Oncol 19 (12): 1602-1616, 2018.
- Gottardo NG, Hansford JR, McGlade JP, et al.: Medulloblastoma Down Under 2013: a report from the third annual meeting of the International Medulloblastoma Working Group. Acta Neuropathol 127 (2): 189-201, 2014.
- Louis DN, Perry A, Burger P, et al.: International Society Of Neuropathology--Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol 24 (5): 429-35, 2014.
- Sharma T, Schwalbe EC, Williamson D, et al.: Second-generation molecular subgrouping of medulloblastoma: an international meta-analysis of Group 3 and Group 4 subtypes. Acta Neuropathol 138 (2): 309-326, 2019.
- von Hoff K, Haberler C, Schmitt-Hoffner F, et al.: Therapeutic implications of improved molecular diagnostics for rare CNS embryonal tumor entities: results of an international, retrospective study. Neuro Oncol 23 (9): 1597-1611, 2021.
- Korshunov A, Sturm D, Ryzhova M, et al.: Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol 128 (2): 279-89, 2014.
- Picard D, Miller S, Hawkins CE, et al.: Markers of survival and metastatic potential in childhood CNS primitive neuro-ectodermal brain tumours: an integrative genomic analysis. Lancet Oncol 13 (8): 838-48, 2012.
- Spence T, Sin-Chan P, Picard D, et al.: CNS-PNETs with C19MC amplification and/or LIN28 expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathol 128 (2): 291-303, 2014.
- Juhnke BO, Gessi M, Gerber NU, et al.: Treatment of embryonal tumors with multilayered rosettes with carboplatin/etoposide induction and high-dose chemotherapy within the prospective P-HIT trial. Neuro Oncol 24 (1): 127-137, 2022.
- Khan S, Solano-Paez P, Suwal T, et al.: Clinical phenotypes and prognostic features of embryonal tumours with multi-layered rosettes: a Rare Brain Tumor Registry study. Lancet Child Adolesc Health 5 (11): 800-813, 2021.
- Kleinman CL, Gerges N, Papillon-Cavanagh S, et al.: Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat Genet 46 (1): 39-44, 2014.
- Li M, Lee KF, Lu Y, et al.: Frequent amplification of a chr19q13.41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell 16 (6): 533-46, 2009.
- Korshunov A, Okonechnikov K, Schmitt-Hoffner F, et al.: Molecular analysis of pediatric CNS-PNET revealed nosologic heterogeneity and potent diagnostic markers for CNS neuroblastoma with FOXR2-activation. Acta Neuropathol Commun 9 (1): 20, 2021.
- Ueno-Yokohata H, Okita H, Nakasato K, et al.: Consistent in-frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney. Nat Genet 47 (8): 861-3, 2015.
- Roy A, Kumar V, Zorman B, et al.: Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat Commun 6: 8891, 2015.
- Louis DN, Ohgaki H, Wiestler OD, et al.: The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114 (2): 97-109, 2007.
- Sharma MC, Mahapatra AK, Gaikwad S, et al.: Pigmented medulloepithelioma: report of a case and review of the literature. Childs Nerv Syst 14 (1-2): 74-8, 1998 Jan-Feb.
- Jakobiec FA, Kool M, Stagner AM, et al.: Intraocular Medulloepitheliomas and Embryonal Tumors With Multilayered Rosettes of the Brain: Comparative Roles of LIN28A and C19MC. Am J Ophthalmol 159 (6): 1065-1074.e1, 2015.
- Korshunov A, Jakobiec FA, Eberhart CG, et al.: Comparative integrated molecular analysis of intraocular medulloepitheliomas and central nervous system embryonal tumors with multilayered rosettes confirms that they are distinct nosologic entities. Neuropathology 35 (6): 538-44, 2015.
- Keck MK, Sill M, Wittmann A, et al.: Amplification of the PLAG-family genes-PLAGL1 and PLAGL2-is a key feature of the novel tumor type CNS embryonal tumor with PLAGL amplification. Acta Neuropathol 145 (1): 49-69, 2023.
- de Jong MC, Kors WA, de Graaf P, et al.: Trilateral retinoblastoma: a systematic review and meta-analysis. Lancet Oncol 15 (10): 1157-67, 2014.
- Ramasubramanian A, Kytasty C, Meadows AT, et al.: Incidence of pineal gland cyst and pineoblastoma in children with retinoblastoma during the chemoreduction era. Am J Ophthalmol 156 (4): 825-9, 2013.
- Abramson DH, Dunkel IJ, Marr BP, et al.: Incidence of pineal gland cyst and pineoblastoma in children with retinoblastoma during the chemoreduction era. Am J Ophthalmol 156 (6): 1319-20, 2013.
- Turaka K, Shields CL, Meadows AT, et al.: Second malignant neoplasms following chemoreduction with carboplatin, etoposide, and vincristine in 245 patients with intraocular retinoblastoma. Pediatr Blood Cancer 59 (1): 121-5, 2012.
- de Kock L, Sabbaghian N, Druker H, et al.: Germ-line and somatic DICER1 mutations in pineoblastoma. Acta Neuropathol 128 (4): 583-95, 2014.
- Liu APY, Li BK, Pfaff E, et al.: Clinical and molecular heterogeneity of pineal parenchymal tumors: a consensus study. Acta Neuropathol 141 (5): 771-785, 2021.
- Pajtler KW, Witt H, Sill M, et al.: Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 27 (5): 728-43, 2015.
- Witt H, Mack SC, Ryzhova M, et al.: Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 20 (2): 143-57, 2011.
- Mack SC, Witt H, Piro RM, et al.: Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506 (7489): 445-50, 2014.
- Pajtler KW, Mack SC, Ramaswamy V, et al.: The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta Neuropathol 133 (1): 5-12, 2017.
- Zschernack V, Jünger ST, Mynarek M, et al.: Supratentorial ependymoma in childhood: more than just RELA or YAP. Acta Neuropathol 141 (3): 455-466, 2021.
- Ramaswamy V, Hielscher T, Mack SC, et al.: Therapeutic Impact of Cytoreductive Surgery and Irradiation of Posterior Fossa Ependymoma in the Molecular Era: A Retrospective Multicohort Analysis. J Clin Oncol 34 (21): 2468-77, 2016.
- Korshunov A, Witt H, Hielscher T, et al.: Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol 28 (19): 3182-90, 2010.
- Merchant TE, Bendel AE, Sabin ND, et al.: Conformal Radiation Therapy for Pediatric Ependymoma, Chemotherapy for Incompletely Resected Ependymoma, and Observation for Completely Resected, Supratentorial Ependymoma. J Clin Oncol 37 (12): 974-983, 2019.
- Baroni LV, Sundaresan L, Heled A, et al.: Ultra high-risk PFA ependymoma is characterized by loss of chromosome 6q. Neuro Oncol 23 (8): 1360-1370, 2021.
- Panwalkar P, Clark J, Ramaswamy V, et al.: Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol 134 (5): 705-714, 2017.
- Chapman RJ, Ghasemi DR, Andreiuolo F, et al.: Optimizing biomarkers for accurate ependymoma diagnosis, prognostication, and stratification within International Clinical Trials: A BIOMECA study. Neuro Oncol 25 (10): 1871-1882, 2023.
- Pajtler KW, Wen J, Sill M, et al.: Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol 136 (2): 211-226, 2018.
- Gessi M, Capper D, Sahm F, et al.: Evidence of H3 K27M mutations in posterior fossa ependymomas. Acta Neuropathol 132 (4): 635-7, 2016.
- Ryall S, Guzman M, Elbabaa SK, et al.: H3 K27M mutations are extremely rare in posterior fossa group A ependymoma. Childs Nerv Syst 33 (7): 1047-1051, 2017.
- Cavalli FMG, Hübner JM, Sharma T, et al.: Heterogeneity within the PF-EPN-B ependymoma subgroup. Acta Neuropathol 136 (2): 227-237, 2018.
- Parker M, Mohankumar KM, Punchihewa C, et al.: C11orf95-RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature 506 (7489): 451-5, 2014.
- Pietsch T, Wohlers I, Goschzik T, et al.: Supratentorial ependymomas of childhood carry C11orf95-RELA fusions leading to pathological activation of the NF-κB signaling pathway. Acta Neuropathol 127 (4): 609-11, 2014.
- Pagès M, Pajtler KW, Puget S, et al.: Diagnostics of pediatric supratentorial RELA ependymomas: integration of information from histopathology, genetics, DNA methylation and imaging. Brain Pathol 29 (3): 325-335, 2019.
- Jünger ST, Andreiuolo F, Mynarek M, et al.: CDKN2A deletion in supratentorial ependymoma with RELA alteration indicates a dismal prognosis: a retrospective analysis of the HIT ependymoma trial cohort. Acta Neuropathol 140 (3): 405-407, 2020.
- Milde T, Pfister S, Korshunov A, et al.: Stepwise accumulation of distinct genomic aberrations in a patient with progressively metastasizing ependymoma. Genes Chromosomes Cancer 48 (3): 229-38, 2009.
- Fukuoka K, Kanemura Y, Shofuda T, et al.: Significance of molecular classification of ependymomas: C11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathol Commun 6 (1): 134, 2018.
- Ghasemi DR, Sill M, Okonechnikov K, et al.: MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol 138 (6): 1075-1089, 2019.
- Swanson AA, Raghunathan A, Jenkins RB, et al.: Spinal Cord Ependymomas With MYCN Amplification Show Aggressive Clinical Behavior. J Neuropathol Exp Neurol 78 (9): 791-797, 2019.
- Scheil S, Brüderlein S, Eicker M, et al.: Low frequency of chromosomal imbalances in anaplastic ependymomas as detected by comparative genomic hybridization. Brain Pathol 11 (2): 133-43, 2001.
- Raffeld M, Abdullaev Z, Pack SD, et al.: High level MYCN amplification and distinct methylation signature define an aggressive subtype of spinal cord ependymoma. Acta Neuropathol Commun 8 (1): 101, 2020.
Liver Cancer
Hepatoblastoma
Molecular features of hepatoblastoma
Genomic findings related to hepatoblastoma include the following:
- The frequency of variants in hepatoblastoma, as determined by three groups using whole-exome sequencing, was very low (approximately three variants per tumor) in children younger than 5 years. A pediatric pan-cancer genomics study found that hepatoblastoma had the lowest gene variant rate among all childhood cancers studied.
- Hepatoblastoma is primarily a disease of WNT pathway activation. The primary mechanism for WNT pathway activation is CTNNB1 activating variants/deletions involving exon 3. CTNNB1 variants have been reported in more than 80% of cases. A less common cause of WNT pathway activation in hepatoblastoma is variants in APC associated with familial adenomatosis polyposis coli.
- NFE2L2 variants were identified in 10 of 174 (6%), 4 of 88 (5%), and 5 of 112 (4%) cases of hepatoblastoma in three studies. The presence of NFE2L2 variants was associated with a lower survival rate.
- Similar NFE2L2 variants have been found in many types of cancer, including hepatocellular carcinoma. These variants render NFE2L2 insensitive to KEAP1-mediated degradation, leading to activation of the NFE2L2-KEAP1 pathway, which activates resistance to oxidative stress and is believed to confer resistance to chemotherapy.
- TERT and TP53 variants, which are common in adults with hepatocellular carcinoma, are uncommon in children with hepatoblastoma. Pediatric patients with TERT variants present with hepatoblastoma at a significantly older age, compared with patients without TERT variants (median age at diagnosis, approximately 10 years vs. 1.4 years).
- Uniparental disomy at 11p15.5 with loss of the maternal allele was reported in 6 of 15 cases of hepatoblastoma. This finding has been confirmed in genomic characterization studies, in which 30% to 40% of cases showed allelic imbalance at the 11p15 locus.
Gene expression and epigenetic profiling have been used to identify biological subtypes of hepatoblastoma and to evaluate the prognostic significance of these subtypes.
- A 16-gene expression signature divided hepatoblastoma cases into two subsets, C1 and C2. The C1 subtype included most of the well-differentiated fetal (pure fetal) histology cases. The C2 subtype showed a more immature pattern and was associated with higher rates of metastatic disease at diagnosis. In a study of 174 patients with hepatoblastoma, the C2 subtype was a significant predictor of poor outcome in multivariable analysis.
- A second research group also found that gene expression profiling could be used to identify subsets of hepatoblastoma with favorable versus unfavorable prognosis. The unfavorable prognosis group of patients showed elevated expression of genes associated with embryonic stem cell and progenitor cells (e.g., LIN28B, SALL4, and HMGA2). The favorable prognosis group of patients showed elevated expression of genes associated with liver differentiation (e.g., HNF1A).
- A gene expression signature at chromosome 14q32 (e.g., DLK1) was identified, with a stronger expression signal being associated with higher risk of treatment failure. A strong 14q32 expression signature was also observed in fetal liver tissue, further supporting the concept that patients with hepatoblastoma who have tumors with biological characteristics that are similar to those of hepatic precursor cells have an inferior prognosis.
- Epigenetic profiling of hepatoblastoma has been used to identify molecularly defined hepatoblastoma subtypes. Tumors from 113 patients with hepatoblastoma were evaluated using DNA methylation arrays. Two distinctive subtypes were identified, epigenetic cluster A and B (Epi-CA and Epi-CB). The methylation profile of Epi-CB resembled that of early embryonal/fetal phases of liver development. The methylation profile of Epi-CA was similar to that of late fetal or postnatal liver phases. Event-free survival was significantly lower for patients with the Epi-CB subtype than for those with the Epi-CA subtype.
Delineating the clinical applications of these genomic, transcriptomic, and epigenomic profiling methods for the risk classification of patients with hepatoblastoma will require independent validation, which is one of the objectives of the Paediatric Hepatic International Tumour Trial (PHITT [NCT03017326]).
Hepatocellular Carcinoma
Molecular features of hepatocellular carcinoma
Genomic findings related to hepatocellular carcinoma include the following:
- One case of pediatric hepatocellular carcinoma was analyzed by whole-exome sequencing, which showed a higher variant rate (53 variants) and the coexistence of CTNNB1 and NFE2L2 variants.
- One study investigated pediatric (nonfibrolamellar) hepatocellular carcinoma tumors (N = 15) using multiple analytic tools. These tumors were found to frequently carry aberrations in a subset of genes that are commonly altered in adult hepatocellular carcinoma, including CTNNB1 and TERT. However, the molecular mechanisms of the variants are different. The TP53 variant was rare in this pediatric hepatocellular carcinoma cohort. Pediatric hepatocellular carcinoma that arose in the background of underlying metabolic disease had fewer variants and a distinct molecular profile. Typical driver variants were lacking in this group of patients.
- A rare, more aggressive subtype of childhood liver cancer (hepatocellular neoplasm, not otherwise specified, also termed transitional liver cell tumor) occurs in older children. It has clinical and histopathological findings of both hepatoblastoma and hepatocellular carcinoma.
TERT variants were observed in two of four transitional liver cell tumor cases tested. TERT variants are also commonly observed in adults with hepatocellular carcinoma.
To date, these genetic variants have not been used to select therapeutic agents for investigation in clinical trials.
Fibrolamellar Carcinoma
Molecular features of fibrolamellar carcinoma
Fibrolamellar carcinoma is a rare subtype of hepatocellular carcinoma observed in older children and young adults. It is characterized by an approximately 400 kB deletion on chromosome 19, which produces a chimeric transcript. This chimeric RNA codes for a protein containing the amino-terminal domain of DNAJB1, a homolog of the molecular chaperone DNAJ, fused in frame with PRKACA, the catalytic domain of protein kinase A.
For information about the treatment of childhood liver cancer, see Childhood Liver Cancer Treatment.
References
- Eichenmüller M, Trippel F, Kreuder M, et al.: The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J Hepatol 61 (6): 1312-20, 2014.
- Jia D, Dong R, Jing Y, et al.: Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology 60 (5): 1686-96, 2014.
- Sumazin P, Chen Y, Treviño LR, et al.: Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology 65 (1): 104-121, 2017.
- Carrillo-Reixach J, Torrens L, Simon-Coma M, et al.: Epigenetic footprint enables molecular risk stratification of hepatoblastoma with clinical implications. J Hepatol 73 (2): 328-341, 2020.
- Gröbner SN, Worst BC, Weischenfeldt J, et al.: The landscape of genomic alterations across childhood cancers. Nature 555 (7696): 321-327, 2018.
- Sekiguchi M, Seki M, Kawai T, et al.: Integrated multiomics analysis of hepatoblastoma unravels its heterogeneity and provides novel druggable targets. NPJ Precis Oncol 4: 20, 2020.
- Cairo S, Armengol C, Maibach R, et al.: A combined clinical and biological risk classification improves prediction of outcome in hepatoblastoma patients. Eur J Cancer 141: 30-39, 2020.
- Cancer Genome Atlas Research Network. Electronic address: [email protected], Cancer Genome Atlas Research Network: Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 169 (7): 1327-1341.e23, 2017.
- Albrecht S, von Schweinitz D, Waha A, et al.: Loss of maternal alleles on chromosome arm 11p in hepatoblastoma. Cancer Res 54 (19): 5041-4, 1994.
- Cairo S, Armengol C, De Reyniès A, et al.: Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 14 (6): 471-84, 2008.
- Vilarinho S, Erson-Omay EZ, Harmanci AS, et al.: Paediatric hepatocellular carcinoma due to somatic CTNNB1 and NFE2L2 mutations in the setting of inherited bi-allelic ABCB11 mutations. J Hepatol 61 (5): 1178-83, 2014.
- Haines K, Sarabia SF, Alvarez KR, et al.: Characterization of pediatric hepatocellular carcinoma reveals genomic heterogeneity and diverse signaling pathway activation. Pediatr Blood Cancer 66 (7): e27745, 2019.
- Nault JC, Mallet M, Pilati C, et al.: High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun 4: 2218, 2013.
- Honeyman JN, Simon EP, Robine N, et al.: Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science 343 (6174): 1010-4, 2014.
Sarcomas
Osteosarcoma
Molecular Features of Osteosarcoma
The genomic landscape of osteosarcoma is distinct from that of other childhood cancers. Compared with many adult cancers, it is characterized by an exceptionally high number of structural variants with a relatively small number of single nucleotide variants.
Key observations regarding the genomic landscape of osteosarcoma include the following:
- The number of structural variants observed for osteosarcoma is high, at more than 200 structural variants per genome. Thus, osteosarcoma has the most chaotic genome among childhood cancers. The Circos plots shown in Figure 7 illustrate the exceptionally high number of intra- and inter-chromosomal translocations that typify osteosarcoma genomes.
 Figure 7. Circos plots of osteosarcoma cases from the National Cancer Institute's Therapeutically Applicable Research to Generate Effective Treatments (TARGET) project. The red lines in the interior circle connect chromosome regions involved in either intra- or inter-chromosomal translocations. Osteosarcoma is distinctive from other childhood cancers because it has a large number of intra- and inter-chromosomal translocations. Credit: National Cancer Institute.
Figure 7. Circos plots of osteosarcoma cases from the National Cancer Institute's Therapeutically Applicable Research to Generate Effective Treatments (TARGET) project. The red lines in the interior circle connect chromosome regions involved in either intra- or inter-chromosomal translocations. Osteosarcoma is distinctive from other childhood cancers because it has a large number of intra- and inter-chromosomal translocations. Credit: National Cancer Institute. - The tumor mutational burden (TMB) for children and adolescents with osteosarcoma is approximately 2 mutations per megabase and is higher than that of some other childhood cancers (e.g., Ewing sarcoma and rhabdoid tumors). However, this rate is well below that for adult cancers such as melanoma and non-small cell lung cancer, which are responsive to checkpoint inhibitors.
- Rather than activating variants in oncogenes and inactivating variants in tumor suppressor genes, as observed in many cancer types, the genomic landscape for osteosarcoma is driven by copy number gain/amplification in chromosome regions that include oncogenes and copy number loss (deletions) in chromosome regions that include tumor suppressor genes. Recurring copy number gains and losses that affect known oncogenes and tumor suppressor genes, respectively, are described below.
Estimates of the frequency of specific genomic alterations in osteosarcoma vary from report to report. This finding could be a result of different definitions being used to define copy number alterations, different methods being used for their detection, or differences in tumor biology across patient populations (e.g., newly diagnosed versus relapsed, localized versus metastatic, or pediatric versus adult).
- Genomic alterations in TP53, leading to loss of TP53 function, are present in most osteosarcoma cases. A distinctive form of TP53 inactivation occurs through structural variations in the first intron of TP53 that lead to disruption of the TP53 gene. Other mechanisms of TP53 inactivation are also observed, including missense and nonsense variants and deletions of the TP53 gene. The combination of these various mechanisms for loss of TP53 function leads to its biallelic inactivation in most cases of osteosarcoma. Because many of the structural variations leading to TP53 inactivation are best detected through whole-genome sequencing, results based on clinical genomic testing panels may show lower rates of TP53 alterations because they do not detect these changes.
- MDM2 amplification, which is another genomic alteration that leads to loss of TP53 function, is observed in a minority of osteosarcoma cases (approximately 5%).
- RB1 is commonly inactivated in osteosarcoma, sometimes by deleterious variants but more commonly by chromosomal deletion of the chromosome 13q14 region that includes RB1.
- Chromosomal deletions involving chromosome 9p21 lead to CDN2A deletion in approximately 20% of osteosarcoma cases.
- Among tumor oncogenes, MYC at chromosome 8q24 shows gain/amplification in approximately 10% of patients. In one study, MYC gain/amplification appeared to be associated with inferior prognosis. In a second study, MYC gain/amplification was enriched in children, compared with adults.
- CCNE1 at chromosome 19q12 is another tumor oncogene that shows gain/amplification in approximately 10% of patients. Other oncogene-containing chromosomal regions showing chromosomal gain/amplification in a minority of osteosarcoma cases include the CDK4-harboring region at chromosome 12q14, the VEGFA- and CCND3-harboring regions at chromosome 6p12, the CCND1-harboring region at chromosome 11q13, and the PDGFRA-, KIT-, and KDR-harboring regions at chromosome 4q12.
- Alternative lengthening of telomeres (ALT) is the telomere maintenance mechanism employed by the majority of osteosarcoma tumors. ATRX inactivating variants and gene deletions are associated with the ALT telomere maintenance mechanism. ATRX genomic alterations are present in a subset of osteosarcoma tumors that use this telomere maintenance mechanism.
- Many of the genomic alterations reported for osteosarcoma tumors at diagnosis do not provide obvious therapeutic targets, as they reflect loss of tumor suppressor genes (e.g., TP53, RB1, PTEN) rather than activation of targetable oncogenes. In addition, there has been limited success across cancer diagnoses in using gains/amplifications of the oncogenes relevant to osteosarcoma to identify patients that may benefit from targeted therapy.
Genetic predisposition to osteosarcoma
Germline variants in several genes are associated with susceptibility to osteosarcoma. Table 5 summarizes the syndromes and associated genes for these conditions. A recent multi-institutional genomic study of more than 1,200 patients with osteosarcoma revealed pathogenic or likely pathogenic germline variants in autosomal dominant cancer-susceptibility genes in 18% of patients. The frequency of these cancer-susceptibility genes was higher in children aged 10 years or younger.
TP53 variants
Variants in TP53 are the most common germline alterations associated with osteosarcoma. Variants in this gene are found in approximately 70% of patients with Li-Fraumeni syndrome (LFS), which is associated with increased risk of osteosarcoma, breast cancer, various brain cancers, soft tissue sarcomas, and other cancers. While rhabdomyosarcoma is the most common sarcoma arising in patients aged 5 years and younger with TP53-associated LFS, osteosarcoma is the most common sarcoma in children and adolescents aged 6 to 19 years. One study observed a high frequency of young patients (age <30 years) with osteosarcoma carrying a known LFS-associated or likely LFS-associated TP53 variant (3.8%) or rare exonic TP53 variant (5.7%), with an overall TP53 variant frequency of 9.5%. Other groups have reported lower rates (3%–7%) of TP53 germline variants in patients with osteosarcoma.
RECQL4 variants
Investigators analyzed whole-exome sequencing from the germline of 4,435 pediatric cancer patients at the St. Jude Children’s Research Hospital and 1,127 patients from the National Cancer Institute's Therapeutically Applicable Research to Generate Effective Treatment (TARGET) database. They identified 24 patients (0.43%) who harbored loss-of-function RECQL4 variants, including 5 of 249 patients (2.0%) with osteosarcoma. These RECQL4 variants were significantly overrepresented in children with osteosarcoma, the cancer most frequently observed in patients with Rothmund-Thomson syndrome, compared with 134,187 noncancer controls in the Genome Aggregation Database (gnomAD v2.1; P = .00087; odds ratio, 7.1; 95% confidence interval, 2.9–17). Nine of the 24 individuals (38%) possessed the same c.1573delT (p.Cys525Alafs) variant located in the highly conserved DNA helicase domain, suggesting that disruption of this domain is central to oncogenesis.
| Syndrome | Description | Location | Gene | Function |
|---|---|---|---|---|
| AML = acute myeloid leukemia; IL-1 = interleukin-1; MDS = myelodysplastic syndrome; RANKL = receptor activator of nuclear factor kappa beta ligand; TNF = tumor necrosis factor. | ||||
| aAdapted from Kansara et al. | ||||
| Bloom syndrome | Rare inherited disorder characterized by short stature and sun-sensitive skin changes. Often presents with a long, narrow face, small lower jaw, large nose, and prominent ears. | 15q26.1 | BLM | DNA helicase |
| Diamond-Blackfan anemia | Inherited pure red cell aplasia. Patients at risk for MDS and AML. Associated with skeletal abnormalities such as abnormal facial features (flat nasal bridge, widely spaced eyes). | Ribosomal proteins | Ribosome production | |
| Li-Fraumeni syndrome | Inherited variant in TP53 gene. Affected family members at increased risk of bone tumors, breast cancer, leukemia, brain tumors, and sarcomas. | 17p13.1 | TP53 | DNA damage response |
| Paget disease | Excessive breakdown of bone with abnormal bone formation and remodeling, resulting in pain from weak, malformed bone. | 18q21-qa22 | LOH18CR1 | IL-1/TNF signaling; RANKL signaling pathway |
| 5q31 | ||||
| 5q35-qter | ||||
| Retinoblastoma | Malignant tumor of the retina. Approximately 66% of patients are diagnosed by age 2 years and 95% of patients by age 3 years. Patients with heritable germ cell variants at greater risk of subsequent neoplasms. | 13q14.2 | RB1 | Cell-cycle checkpoint |
| Rothmund-Thomson syndrome (also called poikiloderma congenitale) | Autosomal recessive condition. Associated with skin findings (atrophy, telangiectasias, pigmentation), sparse hair, cataracts, small stature, and skeletal abnormalities. Increased incidence of osteosarcoma at a younger age. | 8q24.3 | RECQL4 | DNA helicase |
| Werner syndrome | Patients often have short stature and in their early twenties, develop signs of aging, including graying of hair and hardening of skin. Other aging problems such as cataracts, skin ulcers, and atherosclerosis develop later. | 8p12-p11.2 | WRN | DNA helicase; exonuclease activity |
For more information about these genetic syndromes, see the following summaries:
- Genetics of Breast and Gynecologic Cancers (Li-Fraumeni syndrome [LFS]).
- Genetics of Skin Cancer (Bloom syndrome, Rothmund-Thomson syndrome, and Werner syndrome).
For information about the treatment of osteosarcoma, see Osteosarcoma and Undifferentiated Pleomorphic Sarcoma of Bone Treatment.
Ewing Sarcoma
Molecular Features of Ewing Sarcoma
The detection of a translocation involving the EWSR1 gene on chromosome 22 band q12 and any one of a number of partner chromosomes is the key feature in the diagnosis of Ewing sarcoma (see Table 6). The EWSR1 gene is a member of the TET family [TLS/EWS/TAF15] of RNA-binding proteins. The FLI1 gene is a member of the ETS family of DNA-binding genes. Characteristically, the amino terminus of the EWSR1 gene is juxtaposed with the carboxy terminus of the ETS family genes. In most cases (90%), the carboxy terminus is provided by FLI1, a member of the family of transcription factor genes located on chromosome 11 band q24. Other family members that may combine with the EWSR1 gene are ERG, ETV1, ETV4, and FEV. Rarely, FUS, another TET family member, can substitute for EWSR1. Finally, there are a few rare cases in which EWSR1 has translocated with partners that are not members of the ETS family of oncogenes. The significance of these alternate partners is not known.
Besides these consistent aberrations involving the EWSR1 gene at 22q12, additional numerical and structural aberrations have been observed in Ewing sarcoma, including gains of chromosomes 2, 5, 8, 9, 12, and 15; the nonreciprocal translocation t(1;16)(q12;q11.2); and deletions on the short arm of chromosome 6. Trisomy 20 may be associated with a more aggressive subset of Ewing sarcoma.
Three papers have described the genomic landscape of Ewing sarcoma and all show that these tumors have a relatively silent genome, with a paucity of variants in pathways that might be amenable to treatment with novel targeted therapies. These papers identified recurring genomic alterations in several genes:
- STAG2 variants. Variants in STAG2, a member of the cohesin complex, occur in about 15% to 20% of the cases, and the presence of these variants was associated with advanced-stage disease.
- CDKN2A deletions.CDKN2A deletions were noted in 12% to 22% of cases.
- TP53 variants.TP53 variants were identified in about 6% to 7% of cases and the coexistence of STAG2 and TP53 variants was associated with a poor clinical outcome.
- Gain of whole chromosome 8 (trisomy 8). The most frequent chromosomal alteration in Ewing sarcoma is gain of whole chromosome 8 (trisomy 8), occurring in close to 50% of tumors.
- Gain of chromosome 1q and loss of chromosome 16q. Gain of chromosome 1q and loss of chromosome 16q occur in approximately 20% of patients and often occur together. Chromosome 1q gain and chromosome 16q loss were each associated with inferior prognosis, when analyzed as single factors and when co-occurring. These two chromosomal alterations commonly occur together across a range of cancer types, including Ewing sarcoma. Their co-occurrence is likely a result of their derivation from an unbalanced t(1;16) translocation resulting in gain of chromosome 1q together with loss of chromosomal material from 16q.
A discovery cohort (n = 99) highlighted the frequency of chromosome 8 gain, the co-occurrence of chromosome 1q gain and chromosome 16q loss, the mutual exclusivity of CDKN2A deletion and STAG2 variant, and the relative paucity of recurrent single nucleotide variants for Ewing sarcoma.
Ewing sarcoma translocations can all be found with standard cytogenetic analysis. A more rapid analysis looking for a break apart of the EWSR1 gene is now frequently done to confirm the diagnosis of Ewing sarcoma molecularly. This test result must be considered with caution, however. Ewing sarcomas that utilize FUS translocations will have negative tests because the EWSR1 gene is not translocated in those cases. In addition, other small round tumors also contain translocations of different ETS family members with EWSR1, such as desmoplastic small round cell tumor, clear cell sarcoma, extraskeletal myxoid chondrosarcoma, and myxoid liposarcoma, all of which may be positive with a EWSR1 fluorescence in situ hybridization (FISH) break-apart probe. A detailed analysis of 85 patients with small round blue cell tumors that were negative for EWSR1 rearrangement by FISH with an EWSR1 break-apart probe identified eight patients with FUS rearrangements. Four patients who had EWSR1::ERG fusions were not detected by FISH with an EWSR1 break-apart probe. The authors do not recommend relying solely on EWSR1 break-apart probes for analyzing small round blue cell tumors with strong immunohistochemical positivity for CD99.
Genome-wide association studies have identified susceptibility loci for Ewing sarcoma at 1p36.22, 10q21, and 15q15. Deep sequencing through the 10q21.3 region identified a polymorphism in the EGR2 gene, which appears to cooperate with and magnify the enhanced activity of the gene product of the EWSR1::FLI1 fusion that is seen in most patients with Ewing sarcoma. The polymorphism associated with the increased risk is found at a much higher frequency in White people than in Black or Asian people, possibly contributing to the epidemiology of the relative infrequency of Ewing sarcoma in the latter populations. Three new susceptibility loci have been identified at 6p25.1, 20p11.22, and 20p11.23.
| TET Family Partner | Fusion With ETS-Like Oncogene Partner | Translocation | Comment |
|---|---|---|---|
| aThese partners are not members of the ETS family of oncogenes. | |||
| EWSR1 | EWSR1::FLI1 | t(11;22)(q24;q12) | Most common; approximately 85% to 90% of cases |
| EWSR1::ERG | t(21;22)(q22;q12) | Second most common; approximately 10% of cases | |
| EWSR1::ETV1 | t(7;22)(p22;q12) | Rare | |
| EWSR1::ETV4 | t(17;22)(q12;q12) | Rare | |
| EWSR1::FEV | t(2;22)(q35;q12) | Rare | |
| EWSR1::NFATC2a | t(20;22)(q13;q12) | Rare | |
| EWSR1::POU5F1a | t(6;22)(p21;q12) | ||
| EWSR1::SMARCA5a | t(4;22)(q31;q12) | Rare | |
| EWSR1::PATZ1a | t(6;22)(p21;q12) | ||
| EWSR1::SP3a | t(2;22)(q31;q12) | Rare | |
| FUS | FUS::ERG | t(16;21)(p11;q22) | Rare |
| FUS::FEV | t(2;16)(q35;p11) | Rare | |
For information about the treatment of Ewing sarcoma, see Ewing Sarcoma and Undifferentiated Small Round Cell Sarcomas of Bone and Soft Tissue Treatment.
Rhabdomyosarcoma
Genomics of rhabdomyosarcoma
The four histological categories recognized in the 5th edition of the World Health Organization (WHO) Classification of Tumors of Soft Tissue and Bone have distinctive genomic alterations and are briefly summarized below.
- Embryonal rhabdosarcoma: Characterized by loss of heterozygosity at 11p15 and by a high frequency of variants in genes in the RAS pathway. For the purposes of this section, patients with embryonal rhabdomyosarcoma are considered negative for PAX3::FOXO1 and PAX7::FOXO1 gene fusions (i.e., fusion-negative rhabdomyosarcoma).
- Alveolar rhabdomyosarcoma: Characterized by gene fusions involving FOXO1 with either PAX3 or PAX7 (i.e., FOXO1 fusion–positive rhabdomyosarcoma). Cases with alveolar rhabdomyosarcoma histology without FOXO1 gene fusions have clinical behavior, gene alteration patterns, and transcriptomic profiles like cases with embryonal rhabdomyosarcoma. Therefore, the discussion below focuses only on alveolar rhabdomyosarcoma with FOXO1 gene fusions.
- Spindle cell/sclerosing rhabdomyosarcoma: Characterized by variants of MYOD1 in older patients and by VGLL2 and NCOA2 gene rearrangements in young children.
- Pleomorphic rhabdomyosarcoma: Characterized by complex karyotypes with numerical and unbalanced structural changes that are indistinguishable from those of undifferentiated pleomorphic sarcomas.
The distribution of gene variants and gene amplifications (for CDK4 and MYCN) differs between patients with embryonal histology lacking a PAX::FOXO1 gene fusion (fusion-negative rhabdomyosarcoma) and patients with PAX::FOXO1 gene fusions (fusion-positive rhabdomyosarcoma). See Table 7 below and the text that follows. These frequencies are derived from a combined cohort of the Children's Oncology Group (COG) and United Kingdom rhabdomyosarcoma patients (n = 641).
| Gene | % FN Cases With Gene Alteration | % FP Cases With Gene Alteration |
|---|---|---|
| aAdapted from Shern et al. | ||
| NRAS | 17% | 1% |
| KRAS | 9% | 1% |
| HRAS | 8% | 2% |
| FGFR4 | 13% | 0% |
| NF1 | 15% | 4% |
| BCOR | 15% | 6% |
| TP53 | 13% | 4% |
| CTNNB1 | 6% | 0% |
| CDK4 | 0% | 13% |
| MYCN | 0% | 10% |
Details of the genomic alterations that predominate within each of the WHO histological categories are as follows.
- Fusion-negative rhabdomyosarcoma (embryonal histology): Embryonal rhabdomyosarcoma tumors often show loss of heterozygosity at 11p15 and gains on chromosome 8. Embryonal tumors have a higher background variant rate and a higher single-nucleotide variant rate than do alveolar rhabdomyosarcoma tumors, and the number of somatic variants increases with older age at diagnosis. The most common recurring variants include those in the RAS pathway (e.g., NRAS, KRAS, HRAS, and NF1), which together are observed in approximately one-half of cases. Variants in NRAS are the most frequent RAS pathway gene variants beyond infancy, while variants in HRAS predominate during infancy. The presence of a RAS variant does not confer prognostic significance.
Among the RAS pathway genes, germline variants in NF1 and HRAS predispose to rhabdomyosarcoma. In a study of 615 children with rhabdomyosarcoma, 347 had tumors with embryonal histology. Of these, nine patients had NF1 germline variants, and five patients had HRAS germline variants, representing 2.6% and 1.4% of embryonal histology cases, respectively.
Other genes with recurring variants in fusion-negative rhabdomyosarcoma tumors include FGFR4, PIK3CA, CTNNB1, FBXW7, and BCOR, all of which are present in fewer than 15% of cases.
TP53 variants:TP53 variants are observed in 10% to 15% of patients with fusion-negative rhabdomyosarcoma and occur less commonly (about 4%) in patients with alveolar rhabdomyosarcoma. In other childhood cancers (e.g., Wilms tumor), TP53 variants are associated with anaplastic histology, and the same is true for embryonal rhabdomyosarcoma. In a study of 146 rhabdomyosarcoma patients with known TP53 status, approximately two-thirds of tumors with TP53 variants showed anaplasia (69%), but only one-quarter of tumors with anaplasia had TP53 variants.
The presence of TP53 variants was associated with reduced EFS in both nonrisk-stratified and risk-stratified analyses for both a COG and a U.K. rhabdomyosarcoma cohort. The poor prognosis associated with TP53 variants was observed for both embryonal and alveolar patients. Based on these results, the COG plans to consider TP53 variant as a high-risk defining characteristic in its upcoming trials.
Rhabdomyosarcoma is one of the childhood cancers associated with Li-Fraumeni syndrome. In a study of 614 pediatric patients with rhabdomyosarcoma, 11 patients (1.7%) had TP53 germline variants. Variants were less common in patients with alveolar histology (0.6%), compared with patients with nonalveolar histologies (2.2%). Rhabdomyosarcoma with nonalveolar anaplastic morphology may be a presenting feature for children with Li-Fraumeni syndrome and germline TP53 variants.
- Among eight consecutively presenting children with rhabdomyosarcoma and TP53 germline variants, all showed anaplastic morphology. Among an additional seven children with anaplastic rhabdomyosarcoma and unknown TP53 germline variant status, three of the seven children had functionally relevant TP53 germline variants. The median age at diagnosis of the 11 children with TP53 germline variant status was 40 months (range, 19–67 months).
- In another series, 26 of 31 patients with germline TP53 variants had tumors with embryonal histology. Of the 16 tumors that were submitted for central pathology review, 12 had focal or diffuse anaplasia. The median age of patients in this group was 2.3 years.
DICER1 variants in embryonal rhabdomyosarcoma:DICER1 variants are observed in a small subset of patients with embryonal rhabdomyosarcoma, most commonly arising in tumors of the female genitourinary tract. More specifically, most cases of cervical embryonal rhabdomyosarcoma, which most commonly occurs in adolescents and young adults, have DICER1 variants. In contrast, DICER1 variants are rarely observed in patients with vaginal primary sites, an entity occurring primarily in girls younger than 2 or 3 years. DICER1 variants are also common in embryonal rhabdomyosarcoma arising in the uterine corpus, but this presentation is primarily observed in adults. Cervical rhabdomyosarcoma generally shows a sarcoma botryoides histological pattern, and many cases show areas of cartilaginous differentiation, a feature also observed in other tumor types with DICER1 variants. In support of the distinctive biology of embryonal rhabdomyosarcoma with DICER1 variants, these cases have a DNA methylation pattern that is distinctive from that of other embryonal rhabdomyosarcoma cases. A diagnosis of cervical rhabdomyosarcoma is an indication for genetic testing for DICER1 syndrome.
- Fusion-positive rhabdomyosarcoma (alveolar histology): About 70% to 80% of alveolar tumors are characterized by translocations between
the FOXO1 gene on chromosome 13 and either the PAX3 gene on chromosome 2 (t(2;13)(q35;q14)) or the
PAX7 gene on chromosome 1 (t(1;13)(p36;q14)). Other rare fusions include PAX3::NCOA1 and PAX3::INO80D.
Translocations involving the PAX3 gene occur in approximately 60% of alveolar
rhabdomyosarcoma cases, while the PAX7 gene appears to be involved in about 20% of cases. Patients with solid-variant alveolar histology have a lower incidence of PAX::FOXO1 gene fusions than do patients showing classical alveolar histology. The alveolar histology that is associated with the PAX7 gene in patients with or without metastatic disease appears to occur at a younger age and may be associated with longer EFS rates than those associated with PAX3 gene rearrangements. Patients with alveolar histology and the PAX3 gene are older and have a higher incidence of invasive tumor (T2). Around 20% of cases showing alveolar histology have no detectable PAX gene translocation. These patients have clinical behaviors, gene alteration patterns, and transcriptomic profiles that align with patients who have embryonal rhabdomyosarcoma and are now classified together with embryonal rhabdomyosarcoma, as fusion-negative rhabdomyosarcoma.
For the diagnosis of alveolar rhabdomyosarcoma, a FOXO1 gene rearrangement may be detected with good sensitivity and specificity using either fluorescence in situ hybridization or reverse transcription–polymerase chain reaction.
In addition to FOXO1 rearrangements, alveolar tumors are characterized by a lower mutational burden than are fusion-negative tumors, with fewer genes having recurring mutations. The most frequently observed alterations in fusion-positive tumors are focal amplification of CDK4 (13%) or MYCN (10%), with small numbers of patients having recurring mutations in other genes (e.g., BCOR, 6%; NF1, 4%; TP53, 4%; and PIK3CA, 2%). TP53 mutations in alveolar rhabdomyosarcoma appear to connote a high risk of treatment failure.
- Spindle cell/sclerosing histology: Spindle cell/sclerosing rhabdomyosarcoma has been proposed as a separate entity in the WHO Classification of Tumors of Soft Tissue and Bone. Within the spindle cell/sclerosing rhabdomyosarcoma category, several entities have distinctive molecular and clinical characteristics, described below.
Congenital/infantile spindle cell rhabdomyosarcoma: Several reports have described cases of congenital or infantile spindle cell rhabdomyosarcoma with gene fusions involving VGLL2 and NCOA2 (e.g., VGLL2::CITED2, TEAD1::NCOA2, VGLL2::NCOA2, SRF::NCOA2).
- For congenital/infantile spindle cell rhabdomyosarcoma, a study reported that 10 of 11 patients showed recurrent fusion genes. Most of these patients had truncal primary tumors, and there were no paratesticular tumors. Novel VGLL2 rearrangements were observed in seven patients (63%), including the VGLL2::CITED2 fusion in four patients and the VGLL2::NCOA2 fusion in two patients. Three patients (27%) harbored different NCOA2 gene fusions, including TEAD1::NCOA2 in two patients and SRF::NCOA2 in one patient. In this report, all fusion-positive congenital/infantile spindle cell rhabdomyosarcoma patients with long-term follow-up data were alive and well, and no patients developed distant metastases.
- While most studies of congenital/infantile spindle cell rhabdomyosarcoma have shown favorable outcomes, it was reported that four patients developed metastatic disease and two patients had fatal outcomes. Disease progression occurred a median of 3.5 years from diagnosis (range, 1–8 years). All four patients had unresectable tumors and were treated with chemotherapy. However, most literature reported cases in which surgical resection was achieved. At disease progression, a tumor from one patient had a TP53 variant, and a tumor from another patient showed a homozygous CDKN2A and CDKN2B deletion.
- A study of 40 patients with congenital/infantile spindle cell rhabdomyosarcoma (defined by diagnosis at age ≤12 months) found that almost all patients had localized disease (n = 39) and that one-half of patients who underwent molecular testing (13 of 26) had rearrangements of NCOA2 and/or VGLL2. Because testing was limited to NCOA2 and VGLL2, it is possible that more comprehensive genomic analysis would identify a higher proportion of patients with relevant gene fusions. The 5-year EFS rate for the 13 patients with either a VGLL2 and/or a NCOA2 fusion was 90% (95% CI, ±19%), and the overall survival (OS) rate was 100% (95% CI, ±9%).
- Further study is needed to better define the prevalence and prognostic significance of gene rearrangements in VGLL2, NCOA2, and other relevant genes in young children with congenital/infantile spindle cell rhabdomyosarcoma.
MYOD1-altered spindle cell/sclerosing rhabdomyosarcoma: In older children and adults with spindle cell/sclerosing rhabdomyosarcoma, a specific MYOD1 variant (p.L122R) has been observed in a large proportion of patients. In the combined cohort of COG and U.K. rhabdomyosarcoma patients (n = 641), variants in MYOD1 were found in 3% (17 of 515) of all fusion-negative rhabdomyosarcoma cases and in no fusion-positive cases. The presenting age of patients with MYOD1 variants was 10.8 years. Most cases in this cohort showed spindle or sclerosing features, but cases with densely packed cells that mimicked the dense pattern of embryonal rhabdomyosarcoma were also observed. Most cases in this cohort (15 of 17, 88%) had either head and neck or parameningeal region primary sites. Activating PIK3CA variants are seen in about one-half of cases with MYOD1 variants. The presence of the MYOD1 variant is associated with a markedly increased risk of local and distant failure.
Intraosseous spindle cell rhabdomyosarcoma: Primary intraosseous rhabdomyosarcoma is a very uncommon presentation for rhabdomyosarcoma. Most cases present with gene rearrangements involving TFCP2, with either FUS or EWSR1. Rhabdomyosarcoma with a FUS::TFCP2 or EWSR1::TFCP2 gene fusion most commonly presents in young adults, although cases in older children and adolescents have been reported. Craniofacial bones are the most common primary tumor location, and positivity for ALK and cytokeratins by immunohistochemistry is commonly observed. Other characteristics of this entity include a complex genomic profile, with most cases showing deletion of the CDKN2A tumor suppressor gene. Intraosseous spindle cell rhabdomyosarcoma with a FUS::TFCP2 or EWSR1::TFCP2 gene fusion shows an aggressive clinical course. In one study, the median OS was only 8 months.
Recurrent and refractory rhabdomyosarcomas from pediatric (n = 105) and young-adult patients (n = 15) underwent tumor sequencing in the National Cancer Institute–Children's Oncology Group (NCI-COG) Pediatric MATCH trial. Actionable genomic alterations were found in 53 of 120 tumors (44.2%), and patients with these alterations qualified for treatment on MATCH study arms. Variants of MAPK pathway genes (HRAS, KRAS, NRAS, NF1) were most frequent and were reported in 32 of 120 tumors (26.7%). Amplifications of cyclin-dependent kinase genes (CDK4, CDK6) were detected in 15 of 120 tumors (12.5%).
For information about the treatment of childhood rhabdomyosarcoma, see Childhood Rhabdomyosarcoma Treatment.
References
- Chen X, Bahrami A, Pappo A, et al.: Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep 7 (1): 104-12, 2014.
- Perry JA, Kiezun A, Tonzi P, et al.: Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U S A 111 (51): E5564-73, 2014.
- Marinoff AE, Spurr LF, Fong C, et al.: Clinical Targeted Next-Generation Panel Sequencing Reveals MYC Amplification Is a Poor Prognostic Factor in Osteosarcoma. JCO Precis Oncol 7: e2200334, 2023.
- Suehara Y, Alex D, Bowman A, et al.: Clinical Genomic Sequencing of Pediatric and Adult Osteosarcoma Reveals Distinct Molecular Subsets with Potentially Targetable Alterations. Clin Cancer Res 25 (21): 6346-6356, 2019.
- Nacev BA, Sanchez-Vega F, Smith SA, et al.: Clinical sequencing of soft tissue and bone sarcomas delineates diverse genomic landscapes and potential therapeutic targets. Nat Commun 13 (1): 3405, 2022.
- De Noon S, Ijaz J, Coorens TH, et al.: MYC amplifications are common events in childhood osteosarcoma. J Pathol Clin Res 7 (5): 425-431, 2021.
- Parsons DW, Janeway KA, Patton DR, et al.: Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients With Refractory Cancers in the National Cancer Institute-Children's Oncology Group Pediatric MATCH Trial. J Clin Oncol 40 (20): 2224-2234, 2022.
- Sanders RP, Drissi R, Billups CA, et al.: Telomerase expression predicts unfavorable outcome in osteosarcoma. J Clin Oncol 22 (18): 3790-7, 2004.
- de Nonneville A, Salas S, Bertucci F, et al.: TOP3A amplification and ATRX inactivation are mutually exclusive events in pediatric osteosarcomas using ALT. EMBO Mol Med 14 (10): e15859, 2022.
- Mirabello L, Zhu B, Koster R, et al.: Frequency of Pathogenic Germline Variants in Cancer-Susceptibility Genes in Patients With Osteosarcoma. JAMA Oncol 6 (5): 724-734, 2020.
- Ognjanovic S, Olivier M, Bergemann TL, et al.: Sarcomas in TP53 germline mutation carriers: a review of the IARC TP53 database. Cancer 118 (5): 1387-96, 2012.
- Mirabello L, Yeager M, Mai PL, et al.: Germline TP53 variants and susceptibility to osteosarcoma. J Natl Cancer Inst 107 (7): , 2015.
- Toguchida J, Yamaguchi T, Dayton SH, et al.: Prevalence and spectrum of germline mutations of the p53 gene among patients with sarcoma. N Engl J Med 326 (20): 1301-8, 1992.
- McIntyre JF, Smith-Sorensen B, Friend SH, et al.: Germline mutations of the p53 tumor suppressor gene in children with osteosarcoma. J Clin Oncol 12 (5): 925-30, 1994.
- Maciaszek JL, Oak N, Chen W, et al.: Enrichment of heterozygous germline RECQL4 loss-of-function variants in pediatric osteosarcoma. Cold Spring Harb Mol Case Stud 5 (5): , 2019.
- Kansara M, Thomas DM: Molecular pathogenesis of osteosarcoma. DNA Cell Biol 26 (1): 1-18, 2007.
- German J: Bloom's syndrome. XX. The first 100 cancers. Cancer Genet Cytogenet 93 (1): 100-6, 1997.
- Lipton JM, Federman N, Khabbaze Y, et al.: Osteogenic sarcoma associated with Diamond-Blackfan anemia: a report from the Diamond-Blackfan Anemia Registry. J Pediatr Hematol Oncol 23 (1): 39-44, 2001.
- Idol RA, Robledo S, Du HY, et al.: Cells depleted for RPS19, a protein associated with Diamond Blackfan Anemia, show defects in 18S ribosomal RNA synthesis and small ribosomal subunit production. Blood Cells Mol Dis 39 (1): 35-43, 2007 Jul-Aug.
- Li FP, Fraumeni JF, Mulvihill JJ, et al.: A cancer family syndrome in twenty-four kindreds. Cancer Res 48 (18): 5358-62, 1988.
- Grimer RJ, Cannon SR, Taminiau AM, et al.: Osteosarcoma over the age of forty. Eur J Cancer 39 (2): 157-63, 2003.
- Wong FL, Boice JD, Abramson DH, et al.: Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA 278 (15): 1262-7, 1997.
- Wang LL, Gannavarapu A, Kozinetz CA, et al.: Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst 95 (9): 669-74, 2003.
- Hicks MJ, Roth JR, Kozinetz CA, et al.: Clinicopathologic features of osteosarcoma in patients with Rothmund-Thomson syndrome. J Clin Oncol 25 (4): 370-5, 2007.
- Goto M, Miller RW, Ishikawa Y, et al.: Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev 5 (4): 239-46, 1996.
- Delattre O, Zucman J, Melot T, et al.: The Ewing family of tumors--a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 331 (5): 294-9, 1994.
- Urano F, Umezawa A, Yabe H, et al.: Molecular analysis of Ewing's sarcoma: another fusion gene, EWS-E1AF, available for diagnosis. Jpn J Cancer Res 89 (7): 703-11, 1998.
- Hattinger CM, Rumpler S, Strehl S, et al.: Prognostic impact of deletions at 1p36 and numerical aberrations in Ewing tumors. Genes Chromosomes Cancer 24 (3): 243-54, 1999.
- Sankar S, Lessnick SL: Promiscuous partnerships in Ewing's sarcoma. Cancer Genet 204 (7): 351-65, 2011.
- Roberts P, Burchill SA, Brownhill S, et al.: Ploidy and karyotype complexity are powerful prognostic indicators in the Ewing's sarcoma family of tumors: a study by the United Kingdom Cancer Cytogenetics and the Children's Cancer and Leukaemia Group. Genes Chromosomes Cancer 47 (3): 207-20, 2008.
- Tirode F, Surdez D, Ma X, et al.: Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 4 (11): 1342-53, 2014.
- Crompton BD, Stewart C, Taylor-Weiner A, et al.: The genomic landscape of pediatric Ewing sarcoma. Cancer Discov 4 (11): 1326-41, 2014.
- Brohl AS, Solomon DA, Chang W, et al.: The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet 10 (7): e1004475, 2014.
- Mrózek K, Bloomfield CD: Der(16)t(1;16) is a secondary chromosome aberration in at least eighteen different types of human cancer. Genes Chromosomes Cancer 23 (1): 78-80, 1998.
- Mugneret F, Lizard S, Aurias A, et al.: Chromosomes in Ewing's sarcoma. II. Nonrandom additional changes, trisomy 8 and der(16)t(1;16). Cancer Genet Cytogenet 32 (2): 239-45, 1988.
- Hattinger CM, Rumpler S, Ambros IM, et al.: Demonstration of the translocation der(16)t(1;16)(q12;q11.2) in interphase nuclei of Ewing tumors. Genes Chromosomes Cancer 17 (3): 141-50, 1996.
- Monforte-Muñoz H, Lopez-Terrada D, Affendie H, et al.: Documentation of EWS gene rearrangements by fluorescence in-situ hybridization (FISH) in frozen sections of Ewing's sarcoma-peripheral primitive neuroectodermal tumor. Am J Surg Pathol 23 (3): 309-15, 1999.
- Chen S, Deniz K, Sung YS, et al.: Ewing sarcoma with ERG gene rearrangements: A molecular study focusing on the prevalence of FUS-ERG and common pitfalls in detecting EWSR1-ERG fusions by FISH. Genes Chromosomes Cancer 55 (4): 340-9, 2016.
- Postel-Vinay S, Véron AS, Tirode F, et al.: Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet 44 (3): 323-7, 2012.
- Grünewald TG, Bernard V, Gilardi-Hebenstreit P, et al.: Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat Genet 47 (9): 1073-8, 2015.
- Machiela MJ, Grünewald TGP, Surdez D, et al.: Genome-wide association study identifies multiple new loci associated with Ewing sarcoma susceptibility. Nat Commun 9 (1): 3184, 2018.
- Parham DM, Ellison DA: Rhabdomyosarcomas in adults and children: an update. Arch Pathol Lab Med 130 (10): 1454-65, 2006.
- Newton WA, Gehan EA, Webber BL, et al.: Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 76 (6): 1073-85, 1995.
- WHO Classification of Tumours Editorial Board: WHO Classification of Tumours. Volume 3: Soft Tissue and Bone Tumours. 5th ed., IARC Press, 2020.
- Davicioni E, Anderson JR, Buckley JD, et al.: Gene expression profiling for survival prediction in pediatric rhabdomyosarcomas: a report from the children's oncology group. J Clin Oncol 28 (7): 1240-6, 2010.
- Davicioni E, Anderson MJ, Finckenstein FG, et al.: Molecular classification of rhabdomyosarcoma--genotypic and phenotypic determinants of diagnosis: a report from the Children's Oncology Group. Am J Pathol 174 (2): 550-64, 2009.
- Williamson D, Missiaglia E, de Reyniès A, et al.: Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol 28 (13): 2151-8, 2010.
- Davicioni E, Finckenstein FG, Shahbazian V, et al.: Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res 66 (14): 6936-46, 2006.
- Skapek SX, Anderson J, Barr FG, et al.: PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: a children's oncology group report. Pediatr Blood Cancer 60 (9): 1411-7, 2013.
- Shern JF, Selfe J, Izquierdo E, et al.: Genomic Classification and Clinical Outcome in Rhabdomyosarcoma: A Report From an International Consortium. J Clin Oncol 39 (26): 2859-2871, 2021.
- Merlino G, Helman LJ: Rhabdomyosarcoma--working out the pathways. Oncogene 18 (38): 5340-8, 1999.
- Koufos A, Hansen MF, Copeland NG, et al.: Loss of heterozygosity in three embryonal tumours suggests a common pathogenetic mechanism. Nature 316 (6026): 330-4, 1985 Jul 25-31.
- Scrable H, Witte D, Shimada H, et al.: Molecular differential pathology of rhabdomyosarcoma. Genes Chromosomes Cancer 1 (1): 23-35, 1989.
- Shern JF, Chen L, Chmielecki J, et al.: Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov 4 (2): 216-31, 2014.
- Chen X, Stewart E, Shelat AA, et al.: Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell 24 (6): 710-24, 2013.
- Li H, Sisoudiya SD, Martin-Giacalone BA, et al.: Germline Cancer Predisposition Variants in Pediatric Rhabdomyosarcoma: A Report From the Children's Oncology Group. J Natl Cancer Inst 113 (7): 875-883, 2021.
- Ooms AH, Gadd S, Gerhard DS, et al.: Significance of TP53 Mutation in Wilms Tumors with Diffuse Anaplasia: A Report from the Children's Oncology Group. Clin Cancer Res 22 (22): 5582-5591, 2016.
- Shenoy A, Alvarez E, Chi YY, et al.: The prognostic significance of anaplasia in childhood rhabdomyosarcoma: A report from the Children's Oncology Group. Eur J Cancer 143: 127-133, 2021.
- Haduong JH, Heske CM, Allen-Rhoades W, et al.: An update on rhabdomyosarcoma risk stratification and the rationale for current and future Children's Oncology Group clinical trials. Pediatr Blood Cancer 69 (4): e29511, 2022.
- Hettmer S, Archer NM, Somers GR, et al.: Anaplastic rhabdomyosarcoma in TP53 germline mutation carriers. Cancer 120 (7): 1068-75, 2014.
- Pondrom M, Bougeard G, Karanian M, et al.: Rhabdomyosarcoma associated with germline TP53 alteration in children and adolescents: The French experience. Pediatr Blood Cancer 67 (9): e28486, 2020.
- de Kock L, Yoon JY, Apellaniz-Ruiz M, et al.: Significantly greater prevalence of DICER1 alterations in uterine embryonal rhabdomyosarcoma compared to adenosarcoma. Mod Pathol 33 (6): 1207-1219, 2020.
- Apellaniz-Ruiz M, McCluggage WG, Foulkes WD: DICER1-associated embryonal rhabdomyosarcoma and adenosarcoma of the gynecologic tract: Pathology, molecular genetics, and indications for molecular testing. Genes Chromosomes Cancer 60 (3): 217-233, 2021.
- Kommoss FKF, Stichel D, Mora J, et al.: Clinicopathologic and molecular analysis of embryonal rhabdomyosarcoma of the genitourinary tract: evidence for a distinct DICER1-associated subgroup. Mod Pathol 34 (8): 1558-1569, 2021.
- Dehner LP, Jarzembowski JA, Hill DA: Embryonal rhabdomyosarcoma of the uterine cervix: a report of 14 cases and a discussion of its unusual clinicopathological associations. Mod Pathol 25 (4): 602-14, 2012.
- Daya DA, Scully RE: Sarcoma botryoides of the uterine cervix in young women: a clinicopathological study of 13 cases. Gynecol Oncol 29 (3): 290-304, 1988.
- Bennett JA, Ordulu Z, Young RH, et al.: Embryonal rhabdomyosarcoma of the uterine corpus: a clinicopathological and molecular analysis of 21 cases highlighting a frequent association with DICER1 mutations. Mod Pathol 34 (9): 1750-1762, 2021.
- McCluggage WG, Foulkes WD: DICER1-associated sarcomas: towards a unified nomenclature. Mod Pathol 34 (6): 1226-1228, 2021.
- Schultz KAP, Williams GM, Kamihara J, et al.: DICER1 and Associated Conditions: Identification of At-risk Individuals and Recommended Surveillance Strategies. Clin Cancer Res 24 (10): 2251-2261, 2018.
- Barr FG, Smith LM, Lynch JC, et al.: Examination of gene fusion status in archival samples of alveolar rhabdomyosarcoma entered on the Intergroup Rhabdomyosarcoma Study-III trial: a report from the Children's Oncology Group. J Mol Diagn 8 (2): 202-8, 2006.
- Dumont SN, Lazar AJ, Bridge JA, et al.: PAX3/7-FOXO1 fusion status in older rhabdomyosarcoma patient population by fluorescent in situ hybridization. J Cancer Res Clin Oncol 138 (2): 213-20, 2012.
- Parham DM, Qualman SJ, Teot L, et al.: Correlation between histology and PAX/FKHR fusion status in alveolar rhabdomyosarcoma: a report from the Children's Oncology Group. Am J Surg Pathol 31 (6): 895-901, 2007.
- Sorensen PH, Lynch JC, Qualman SJ, et al.: PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children's oncology group. J Clin Oncol 20 (11): 2672-9, 2002.
- Krsková L, Mrhalová M, Sumerauer D, et al.: Rhabdomyosarcoma: molecular diagnostics of patients classified by morphology and immunohistochemistry with emphasis on bone marrow and purged peripheral blood progenitor cells involvement. Virchows Arch 448 (4): 449-58, 2006.
- Kelly KM, Womer RB, Sorensen PH, et al.: Common and variant gene fusions predict distinct clinical phenotypes in rhabdomyosarcoma. J Clin Oncol 15 (5): 1831-6, 1997.
- Barr FG, Qualman SJ, Macris MH, et al.: Genetic heterogeneity in the alveolar rhabdomyosarcoma subset without typical gene fusions. Cancer Res 62 (16): 4704-10, 2002.
- Missiaglia E, Williamson D, Chisholm J, et al.: PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol 30 (14): 1670-7, 2012.
- Duan F, Smith LM, Gustafson DM, et al.: Genomic and clinical analysis of fusion gene amplification in rhabdomyosarcoma: a report from the Children's Oncology Group. Genes Chromosomes Cancer 51 (7): 662-74, 2012.
- Thway K, Wang J, Wren D, et al.: The comparative utility of fluorescence in situ hybridization and reverse transcription-polymerase chain reaction in the diagnosis of alveolar rhabdomyosarcoma. Virchows Arch 467 (2): 217-24, 2015.
- Nascimento AF, Barr FG: Spindle cell/sclerosing rhabdomyosarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn P, et al., eds.: WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. IARC Press, 2013, pp 134-5.
- Alaggio R, Zhang L, Sung YS, et al.: A Molecular Study of Pediatric Spindle and Sclerosing Rhabdomyosarcoma: Identification of Novel and Recurrent VGLL2-related Fusions in Infantile Cases. Am J Surg Pathol 40 (2): 224-35, 2016.
- Mosquera JM, Sboner A, Zhang L, et al.: Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes Chromosomes Cancer 52 (6): 538-50, 2013.
- Cyrta J, Gauthier A, Karanian M, et al.: Infantile Rhabdomyosarcomas With VGLL2 Rearrangement Are Not Always an Indolent Disease: A Study of 4 Aggressive Cases With Clinical, Pathologic, Molecular, and Radiologic Findings. Am J Surg Pathol 45 (6): 854-867, 2021.
- Whittle S, Venkatramani R, Schönstein A, et al.: Congenital spindle cell rhabdomyosarcoma: An international cooperative analysis. Eur J Cancer 168: 56-64, 2022.
- Kohsaka S, Shukla N, Ameur N, et al.: A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet 46 (6): 595-600, 2014.
- Agaram NP, Chen CL, Zhang L, et al.: Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: evidence for a common pathogenesis. Genes Chromosomes Cancer 53 (9): 779-87, 2014.
- Szuhai K, de Jong D, Leung WY, et al.: Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. J Pathol 232 (3): 300-7, 2014.
- Agaram NP, LaQuaglia MP, Alaggio R, et al.: MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: an aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod Pathol 32 (1): 27-36, 2019.
- Watson S, Perrin V, Guillemot D, et al.: Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol 245 (1): 29-40, 2018.
- Dashti NK, Wehrs RN, Thomas BC, et al.: Spindle cell rhabdomyosarcoma of bone with FUS-TFCP2 fusion: confirmation of a very recently described rhabdomyosarcoma subtype. Histopathology 73 (3): 514-520, 2018.
- Agaram NP, Zhang L, Sung YS, et al.: Expanding the Spectrum of Intraosseous Rhabdomyosarcoma: Correlation Between 2 Distinct Gene Fusions and Phenotype. Am J Surg Pathol 43 (5): 695-702, 2019.
- Le Loarer F, Cleven AHG, Bouvier C, et al.: A subset of epithelioid and spindle cell rhabdomyosarcomas is associated with TFCP2 fusions and common ALK upregulation. Mod Pathol 33 (3): 404-419, 2020.
- Xu B, Suurmeijer AJH, Agaram NP, et al.: Head and neck rhabdomyosarcoma with TFCP2 fusions and ALK overexpression: a clinicopathological and molecular analysis of 11 cases. Histopathology 79 (3): 347-357, 2021.
Langerhans Cell Histiocytosis
Genomics of LCH
BRAF, NRAS, and ARAF variants
The genomic basis of LCH was advanced by a 2010 report of an activating variant of the BRAF oncogene (V600E) that was detected in 35 of 61 cases (57%). Multiple subsequent reports have confirmed the presence of BRAF V600E variants in 50% or more of LCH cases in children. Other BRAF variants that result in signal activation have been described. ARAF variants are infrequent in LCH but, when present, can also lead to RAS-MAPK pathway activation.
The presence of the BRAF V600E variant in blood and bone marrow was studied in a series of 100 patients, 65% of whom tested positive for the BRAF V600E variant by a sensitive quantitative polymerase chain reaction technique. Circulating cells with the BRAF V600E variant could be detected in all high-risk patients and in a subset of low-risk multisystem patients. The BRAF V600E allele was detected in circulating cell-free DNA in 100% of patients with risk-organ–positive multisystem LCH, 42% of patients with risk-organ–negative LCH, and 14% of patients with single-system LCH.
The myeloid dendritic cell origin of LCH was confirmed by finding CD34-positive stem cells with the variant in the bone marrow of high-risk patients. In those with low-risk disease, the variant was found in more mature myeloid dendritic cells, suggesting that the stage of cell development at which the somatic variant occurs is critical in defining the extent of disease in LCH.
Pulmonary LCH in adults was initially reported to be nonclonal in approximately 75% of cases, while a later study of BRAF variants showed that 25% to 50% of adult patients with lung LCH had evidence of BRAF V600E variants. Another study of 26 pulmonary LCH cases found that 50% had BRAF V600E variants and 40% had NRAS variants. Approximately the same number of variants are polyclonal as are monoclonal. It has not been determined whether clonality and BRAF pathway variants are concordant in the same patients, which might suggest a reactive rather than a neoplastic condition in smoker's lung LCH and a clonal neoplasm in other types of LCH.
In a study of 117 patients with LCH, 83 adult patients with pulmonary LCH underwent molecular analysis. Nearly 90% of these patients had variants in the MAPK pathway.[Level of evidence C3] Of the 69 patients who had their biopsy samples further analyzed using a next-generation sequencing panel of 74 genes, 36% had BRAF V600E variants, 29% had BRAF N486-P490 deletions, 15% had MAP2K1 variants or deletions, and 4% had NRAS variants. Only one patient had a KRAS variant. Additionally, 11 patients had their biopsy samples analyzed using whole-exome sequencing. An average of 14 variants were found per patient, which is markedly higher than the average of one variant found per pediatric patient. There were no clinical correlates, including presence of a BRAF V600E variant and smoking status. Of the 117 patients with LCH, 60% experienced a relapse.
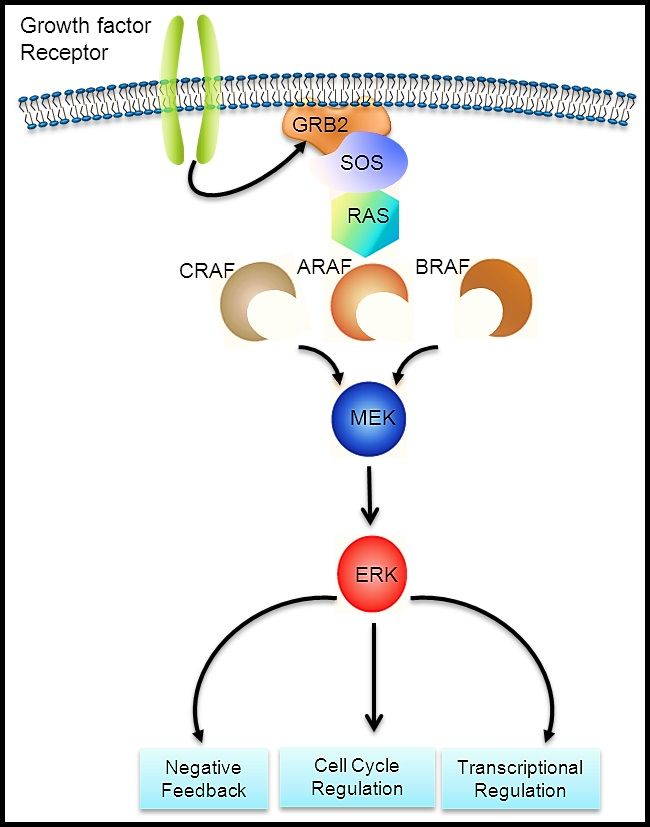 Figure 8. Courtesy of Rikhia Chakraborty, Ph.D. Permission to reuse the figure in any form must be obtained directly from Dr. Chakraborty.
Figure 8. Courtesy of Rikhia Chakraborty, Ph.D. Permission to reuse the figure in any form must be obtained directly from Dr. Chakraborty.
The RAS-MAPK signaling pathway (see Figure 8) transmits signals from a cell surface receptor (e.g., a growth factor) through the RAS pathway (via one of the RAF proteins [A, B, or C]) to phosphorylate MEK and then the extracellular signal-regulated kinase (ERK), which leads to nuclear signals affecting cell cycle and transcription regulation. The V600E variant of BRAF leads to continuous phosphorylation, and thus activation, of MEK and ERK without the need for an external signal. Activation of ERK occurs by phosphorylation, and phosphorylated ERK can be detected in virtually all LCH lesions.
In a mouse model of LCH, the BRAF V600E variant was shown to inhibit a chemokine receptor (CCR7)–mediated migration of dendritic cells, forcing them to accumulate in the LCH lesion. This variant also causes an increased expression of BCL2L1, which results in resistance to apoptosis. This process leads to the cells being less responsive to chemotherapy. The BRAF V600E variant also causes growth arrest of hematopoietic progenitor cells and a senescence-associated secretory phenotype that further promotes accumulation of the pathological cells.
Another mouse model with the BRAF V600E variant under control of Scl or Map17 gene promoters added additional insights into the biology of neurodegenerative LCH. These studies confirmed the hematopoietic origin of CD11a-positive macrophages with BRAF V600E variants. This process disrupts the blood-brain barrier and causes loss of Purkinje cells and progressive neurodegeneration by resistance to apoptosis and production of senescent associated secretory proteins, which include inflammatory cytokines IL-1, IL-6, and matrix metalloproteinases. Treatment with a MAP kinase inhibitor and a senolytic agent (navitoclax) decreased the pathogenic cell numbers and led to clinical improvement in the mice.
In summary, LCH is now considered a myeloid neoplasm primarily driven by activating variants of the MAPK pathway. Fifty percent to 60% of the activating variants are caused by BRAF V600E variants, which are enriched in patients with multisystem risk organ–positive LCH and in patients with neurodegenerative-disease LCH. Ongoing studies are assessing whether low-level variant detection in peripheral blood can be used as a minimal residual disease marker to assist in therapeutic decisions.
Other RAS-MAPK pathway alterations
Because RAS-MAPK pathway activation (elevated phosphor-ERK) can be detected in all LCH cases, including those without BRAF variants, the presence of genomic alterations in other components of the pathway was suspected. The following genomic alterations were identified:
- MAP2K1 variants. Whole-exome sequencing on biopsy samples of BRAF-altered versus BRAF–wild-type LCH tissue revealed that 7 of 21 BRAF–wild-type specimens had MAP2K1 variants, while no BRAF-altered specimens had MAP2K1 variants. The variants in MAP2K1 (which codes for MEK1) were activating, as indicated by their induction of ERK phosphorylation.
Another study showed MAP2K1 variants exclusively in 11 of 22 BRAF–wild-type cases. One study showed that MAP2K1 and other variants associated with pediatric and adult LCH were mutually exclusive of BRAF variants. The authors found a variety of variants in other pathways (e.g., JNK, RAS-ERK, and JAK-STAT) in pediatric and adult patients with BRAF V600E or MAP2K1 variants. Another study evaluated the kinase alterations and myeloid-associated variants in 73 adult patients with LCH. They reported a median of two variants per adult patient, as opposed to children who usually have only one variant. BRAF V600E was found in 31%, BRAF indel in 29%, and MAP2K1 in 19% of patients with LCH. A variety of other protein kinase and related pathways were found in 89% of adult patients with LCH. MAP2K1 variants were exclusive of BRAF variants.
- In-frame BRAF deletions and FAM73A::BRAF gene fusions. In-frame BRAF deletions and in-frame FAM73A::BRAF gene fusions have occurred in the group of BRAF V600E and MAP2K1 variant–negative cases.
In summary, studies support the universal activation of ERK in LCH. ERK activation in most cases of LCH is explained by BRAF and MAP2K1 alterations. Altogether, these variants in the MAP kinase pathway account for nearly 80% of the causes of the universal activation of ERK in LCH. The remaining cases have a range of variants that include small deletions in BRAF, BRAF gene fusions (discussed above), as well as variants in ARAF, MAP3K1, NRAS, ERBB3, PI3CA, CSF1R, and other rare targets. [Level of evidence C1]
Clinical implications
Clinical implications of the described genomic findings include the following:
- LCH is included in a group of other pediatric tumors with activating BRAF variants, such as select nonmalignant conditions (e.g., benign nevi) and low-grade malignancies (e.g., pilocytic astrocytoma). All of these conditions have a generally indolent course, with spontaneous resolution occurring in some cases. This distinctive clinical course may be a manifestation of oncogene-induced senescence.
- In some pediatric studies, BRAF V600E variants have been associated with more severe multisystem disease, treatment failure, increased reactivations, and an increased risk of neurodegeneration (see below). These clinical correlates were recently investigated for non-BRAF V600E variants in an international study. Similar to the BRAF V600E cohort, all patients with multisystem risk organ–positive LCH had detectable variants in peripheral blood mononuclear cells. Of seven patients with multisystem risk organ–negative LCH, four had detectable variants. No patients with single-system disease had detectable variants in peripheral blood mononuclear cells. The authors concluded that other MAPK pathway variants are associated with risk status, similar to BRAF V600E variants.
BRAF V600E variants can be targeted by BRAF inhibitors (e.g., vemurafenib and dabrafenib) or by the combination of BRAF inhibitors plus MEK inhibitors (e.g., dabrafenib/trametinib and vemurafenib/cobimetinib). These agents and combinations are approved for adults with melanoma. Treatment of melanoma in adults with combinations of a BRAF inhibitor and a MEK inhibitor showed significantly improved progression-free survival outcomes compared with treatment using a BRAF inhibitor alone.
Several case reports and two case series have also demonstrated the efficacy of BRAF inhibitors for the treatment of LCH in children. However, the long-term role of this therapy is complicated because most patients will relapse when the inhibitors are discontinued. For more information, see the sections on Treatment of recurrent, refractory, or progressive high-risk disease: multisystem LCH and Targeted therapies for the treatment of single-system and multisystem disease.
- Circulating BRAF V600E–altered cells have been found in 59% of patients who developed neurodegenerative-disease LCH, compared with 15% of patients who did not develop neurodegenerative-disease LCH. Detectable altered circulating cells had a sensitivity of 0.59 and specificity of 0.86 for developing the neurodegenerative disease. Even after therapy, some patients with neurodegenerative-disease LCH had circulating BRAF V600E–altered cells.
- With additional research, the observation of the BRAF V600E variant (or potentially MAP2K1 variants) in circulating cells or cell-free DNA may become a useful diagnostic tool to define high-risk versus low-risk disease. Additionally, for patients who have a somatic variant, persistence of circulating cells with the variant may be useful as a marker of residual disease.
For information about the treatment of childhood LCH, see Langerhans Cell Histiocytosis Treatment.
References
- Badalian-Very G, Vergilio JA, Degar BA, et al.: Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 116 (11): 1919-23, 2010.
- Berres ML, Lim KP, Peters T, et al.: BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 211 (4): 669-83, 2014.
- Satoh T, Smith A, Sarde A, et al.: B-RAF mutant alleles associated with Langerhans cell histiocytosis, a granulomatous pediatric disease. PLoS One 7 (4): e33891, 2012.
- Sahm F, Capper D, Preusser M, et al.: BRAFV600E mutant protein is expressed in cells of variable maturation in Langerhans cell histiocytosis. Blood 120 (12): e28-34, 2012.
- Héritier S, Hélias-Rodzewicz Z, Chakraborty R, et al.: New somatic BRAF splicing mutation in Langerhans cell histiocytosis. Mol Cancer 16 (1): 115, 2017.
- Nelson DS, Quispel W, Badalian-Very G, et al.: Somatic activating ARAF mutations in Langerhans cell histiocytosis. Blood 123 (20): 3152-5, 2014.
- Héritier S, Hélias-Rodzewicz Z, Lapillonne H, et al.: Circulating cell-free BRAF(V600E) as a biomarker in children with Langerhans cell histiocytosis. Br J Haematol 178 (3): 457-467, 2017.
- Dacic S, Trusky C, Bakker A, et al.: Genotypic analysis of pulmonary Langerhans cell histiocytosis. Hum Pathol 34 (12): 1345-9, 2003.
- Roden AC, Hu X, Kip S, et al.: BRAF V600E expression in Langerhans cell histiocytosis: clinical and immunohistochemical study on 25 pulmonary and 54 extrapulmonary cases. Am J Surg Pathol 38 (4): 548-51, 2014.
- Mourah S, How-Kit A, Meignin V, et al.: Recurrent NRAS mutations in pulmonary Langerhans cell histiocytosis. Eur Respir J 47 (6): 1785-96, 2016.
- Jouenne F, Chevret S, Bugnet E, et al.: Genetic landscape of adult Langerhans cell histiocytosis with lung involvement. Eur Respir J 55 (2): , 2020.
- Chakraborty R, Burke TM, Hampton OA, et al.: Alternative genetic mechanisms of BRAF activation in Langerhans cell histiocytosis. Blood 128 (21): 2533-2537, 2016.
- Chakraborty R, Hampton OA, Shen X, et al.: Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood 124 (19): 3007-15, 2014.
- Hogstad B, Berres ML, Chakraborty R, et al.: RAF/MEK/extracellular signal-related kinase pathway suppresses dendritic cell migration and traps dendritic cells in Langerhans cell histiocytosis lesions. J Exp Med 215 (1): 319-336, 2018.
- Bigenwald C, Le Berichel J, Wilk CM, et al.: BRAFV600E-induced senescence drives Langerhans cell histiocytosis pathophysiology. Nat Med 27 (5): 851-861, 2021.
- Wilk CM, Cathomas F, Török O, et al.: Circulating senescent myeloid cells infiltrate the brain and cause neurodegeneration in histiocytic disorders. Immunity 56 (12): 2790-2802.e6, 2023.
- Milne P, Abhyankar H, Scull B, et al.: Cellular distribution of mutations and association with disease risk in Langerhans cell histiocytosis without BRAFV600E. Blood Adv 6 (16): 4901-4904, 2022.
- Brown NA, Furtado LV, Betz BL, et al.: High prevalence of somatic MAP2K1 mutations in BRAF V600E-negative Langerhans cell histiocytosis. Blood 124 (10): 1655-8, 2014.
- Durham BH, Lopez Rodrigo E, Picarsic J, et al.: Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med 25 (12): 1839-1842, 2019.
- Chen J, Zhao AL, Duan MH, et al.: Diverse kinase alterations and myeloid-associated mutations in adult histiocytosis. Leukemia 36 (2): 573-576, 2022.
- Michaloglou C, Vredeveld LC, Soengas MS, et al.: BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436 (7051): 720-4, 2005.
- Jones DT, Kocialkowski S, Liu L, et al.: Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68 (21): 8673-7, 2008.
- Pfister S, Janzarik WG, Remke M, et al.: BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 118 (5): 1739-49, 2008.
- Jacob K, Quang-Khuong DA, Jones DT, et al.: Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res 17 (14): 4650-60, 2011.
- Héritier S, Emile JF, Barkaoui MA, et al.: BRAF Mutation Correlates With High-Risk Langerhans Cell Histiocytosis and Increased Resistance to First-Line Therapy. J Clin Oncol 34 (25): 3023-30, 2016.
- Larkin J, Ascierto PA, Dréno B, et al.: Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 371 (20): 1867-76, 2014.
- Long GV, Stroyakovskiy D, Gogas H, et al.: Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 386 (9992): 444-51, 2015.
- Eckstein OS, Visser J, Rodriguez-Galindo C, et al.: Clinical responses and persistent BRAF V600E+ blood cells in children with LCH treated with MAPK pathway inhibition. Blood 133 (15): 1691-1694, 2019.
- Donadieu J, Larabi IA, Tardieu M, et al.: Vemurafenib for Refractory Multisystem Langerhans Cell Histiocytosis in Children: An International Observational Study. J Clin Oncol 37 (31): 2857-2865, 2019.
- Kolenová A, Schwentner R, Jug G, et al.: Targeted inhibition of the MAPK pathway: emerging salvage option for progressive life-threatening multisystem LCH. Blood Adv 1 (6): 352-356, 2017.
- Lee LH, Gasilina A, Roychoudhury J, et al.: Real-time genomic profiling of histiocytoses identifies early-kinase domain BRAF alterations while improving treatment outcomes. JCI Insight 2 (3): e89473, 2017.
- Héritier S, Jehanne M, Leverger G, et al.: Vemurafenib Use in an Infant for High-Risk Langerhans Cell Histiocytosis. JAMA Oncol 1 (6): 836-8, 2015.
- Váradi Z, Bánusz R, Csomor J, et al.: Effective BRAF inhibitor vemurafenib therapy in a 2-year-old patient with sequentially diagnosed Langerhans cell histiocytosis and Erdheim-Chester disease. Onco Targets Ther 10: 521-526, 2017.
- McClain KL, Picarsic J, Chakraborty R, et al.: CNS Langerhans cell histiocytosis: Common hematopoietic origin for LCH-associated neurodegeneration and mass lesions. Cancer 124 (12): 2607-2620, 2018.
Neuroblastoma
Molecular features of neuroblastoma
Children with neuroblastoma can be divided into subsets with different predicted risks of relapse on the basis of clinical factors and biological markers at the time of diagnosis.
- Low-risk or intermediate-risk neuroblastoma patients. Patients classified as low risk or intermediate risk have a favorable prognosis, with survival rates exceeding 95%. Low-risk and intermediate-risk neuroblastoma usually occur in children younger than 18 months. These tumors commonly have gains of whole chromosomes and are hyperdiploid when examined by flow cytometry.
- High-risk neuroblastoma patients. The prognosis is more guarded for patients with high-risk neuroblastoma, with a long-term survival rate of less than 50%. High-risk neuroblastoma generally occurs in children older than 18 months and is often metastatic to bone and bone marrow. Segmental chromosome abnormalities (gains or losses) and/or MYCN gene amplification are usually detected in these tumors. They are near diploid or near tetraploid by flow cytometric measurement. High-risk tumors may rarely harbor exonic variants, but most high-risk tumors lack such gene variants. For more information, see the Exonic Variants in Neuroblastoma section.
Key genomic characteristics of high-risk neuroblastoma that are discussed below include the following:
- Segmental chromosomal aberrations.
- MYCN gene amplifications.
- FOXR2 activation.
- Low rates of exonic variants, with activating variants in ALK being the most common recurring alteration.
- Genomic alterations that promote telomere maintenance.
Segmental chromosomal aberrations
Segmental chromosomal aberrations, found most frequently in 1p, 2p, 1q, 3p, 11q, 14q, and 17p, are best detected by comparative genomic hybridization. These aberrations are seen in most high-risk and/or stage 4 neuroblastoma tumors. Among all patients with neuroblastoma, a higher number of chromosome breakpoints (i.e., a higher number of segmental chromosome aberrations) correlated with the following:[Level of evidence C2]
- Advanced age at diagnosis.
- Advanced stage of disease.
- Higher risk of relapse.
- Poorer outcome.
In an analysis of localized, resectable, non-MYCN amplified neuroblastoma, cases from two consecutive European studies and a North American cohort (including INSS stages 1, 2A, and 2B) were analyzed for segmental chromosome aberrations (namely gain of 1q, 2p, and 17q and loss of 1p, 3p, 4p, and 11q). The study revealed a different prognostic impact of tumor genomics depending on patient age (<18 months or >18 months). Patients were treated with surgery alone regardless of a tumor residuum.[Level of evidence C1]
- The presence of segmental chromosome aberrations, especially 11q loss, significantly reduced survival in patients older than 18 months with stage 2 neuroblastoma but not in the cohort of patients younger than 18 months.
- Chromosome 1p loss is a risk factor for relapse but not for diminished overall survival (OS) in patients younger than 18 months. The 5-year event-free survival (EFS) rate was 62% for patients with 1p loss and 87% for patients with no 1p loss (P = .019). The 5-year OS rate was 92% for patients with 1p loss and 97% for patients with no 1p loss.
- Segmental chromosome aberrations (especially 11q loss) are risk factors for reduced EFS and OS in patients older than 18 months. In patients younger than 18 months, only segmental chromosome aberrations led to relapse and death, with 11q loss as the strongest marker (11q loss: 5-year EFS rate, 48%; no 11q loss: 5-year EFS rate, 85%; P = .033; 11q loss: 5-year OS rate, 46%; no 11q loss: 5-year OS rate, 92%; P = .038).
In a study of children older than 12 months who had unresectable primary neuroblastomas without metastases, segmental chromosomal aberrations were found in most patients. Older children were more likely to have them and to have more of them per tumor cell. In children aged 12 to 18 months, the presence of segmental chromosomal aberrations had a significant effect on EFS but not on OS. However, in children older than 18 months, there was a significant difference in OS between children with segmental chromosomal aberrations (67%) and children without segmental chromosomal aberrations (100%), regardless of tumor histology.
Segmental chromosomal aberrations are also predictive of recurrence in infants with localized unresectable or metastatic neuroblastoma without MYCN gene amplification. An analysis of 133 patients (aged ≥18 months) with INSS stage 3 tumors without MYCN amplification demonstrated that segmental chromosomal aberrations were associated with inferior EFS, and 11q loss was independently associated with worse OS.
In an analysis of intermediate-risk patients in a Children's Oncology Group (COG) study, 11q loss, but not 1p loss, was associated with reduced EFS but not OS (11q loss and no 11q loss: 3-year EFS rates, 68% and 85%, respectively; P = .022; 3-year OS rates, 88% and 94%, respectively; P = .09).[Level of evidence B4]
In a multivariable analysis of 407 patients from four consecutive COG high-risk trials, 11q loss of heterozygosity was shown to be a significant predictor of progressive disease, and lack of 11q loss of heterozygosity was associated with both higher rates of end-induction complete response and end-induction partial response.[Level of evidence C1]
An international collaboration studied 556 patients with high-risk neuroblastoma and identified two types of segmental copy number aberrations that were associated with extremely poor outcome. Distal 6q losses were found in 6% of patients and were associated with a 10-year survival rate of only 3.4%. Amplifications of regions not encompassing the MYCN locus, in addition to MYCN amplification, were detected in 18% of the patients and were associated with a 10-year survival rate of 5.8%.
MYCN gene amplification
MYCN amplification is detected in 16% to 25% of neuroblastoma tumors. Among patients with high-risk neuroblastoma, 40% to 50% of cases show MYCN amplification.
In all stages of disease, amplification of the MYCN gene strongly predicts a poorer prognosis, in both time to tumor progression and OS, in almost all multivariate regression analyses of prognostic factors. In the ANBL00B1 (NCT00904241) study of 4,832 newly diagnosed patients enrolled between 2007 to 2017, the 5-year EFS and OS rates were 77% and 87%, respectively, for patients whose tumors were MYCN nonamplified (n = 3,647; 81%). In comparison, the 5-year EFS and OS rates were 51% and 57%, respectively, for patients whose tumors were MYCN amplified (n = 827; 19%).
Within the localized-tumor MYCN-amplified cohort, patients with hyperdiploid tumors have better outcomes than do patients with diploid tumors. However, patients with hyperdiploid tumors with MYCN amplification or any segmental chromosomal aberrations do relatively poorly, compared with patients with hyperdiploid tumors without MYCN amplification.
Most unfavorable clinical and pathobiological features are associated, to some degree, with MYCN amplification. In a multivariable logistic regression analysis of 7,102 patients in the International Neuroblastoma Risk Group (INRG) study, pooled segmental chromosomal aberrations and gains of 17q were poor prognostic features, even when not associated with MYCN amplification. However, another poor prognostic feature, segmental chromosomal aberrations at 11q, are almost entirely mutually exclusive of MYCN amplification.
In a cohort of 6,223 patients from the INRG database with known MYCN status, the OS hazard ratio (HR) associated with MYCN amplification was 6.3 (95% confidence interval [CI], 5.7–7.0; P< .001). The greatest adverse prognostic impact of MYCN amplification for OS was in the youngest patients (aged <18 months: HR, 19.6; aged ≥18 months: HR, 3.0). Patients whose outcome was most impacted by MYCN status were those with otherwise favorable features, including age younger than 18 months, high mitosis-karyorrhexis index, and low ferritin.[Level of evidence C1]
Intratumoral heterogeneous MYCN amplification (hetMNA) refers to the coexistence of MYCN-amplified cells as a cluster or as single scattered cells and non-MYCN–amplified tumor cells. HetMNA has been reported infrequently. It can occur spatially within the tumor as well as between the tumor and the metastasis at the same time or temporally during the disease course. The International Society of Paediatric Oncology Europe Neuroblastoma (SIOPEN) biology group investigated the prognostic significance of this neuroblastoma subtype. Tumor tissue from 99 patients identified as having hetMNA and diagnosed between 1991 and 2015 was analyzed to elucidate the prognostic significance of MYCN-amplified clones in otherwise non-MYCN–amplified neuroblastomas. Patients younger than 18 months showed a better outcome in all stages compared with older patients. The genomic background correlated significantly with relapse frequency and OS. No relapses occurred in cases of only numerical chromosomal aberrations. This study suggests that hetMNA tumors be evaluated in the context of the genomic tumor background in combination with the clinical pattern, including the patient's age and disease stage. Future studies are needed in patients younger than 18 months who have localized disease with hetMNA.
FOXR2 activation
FOXR2 gene expression is observed in approximately 8% of neuroblastoma cases. FOXR2 gene expression is normally absent postnatally, with the exception of male reproductive tissues. FOXR2 expression is also observed in a subset of central nervous system (CNS) primitive neuroectodermal tumors, termed CNS NB-FOXR2. FOXR2 overexpression was virtually mutually exclusive in neuroblastoma tumors with both elevated MYC and MYCN expression. Although MYCN gene expression was not elevated in neuroblastoma with FOXR2 activation, the gene expression profile for the FOXR2 expressing cases closely resembled that of MYCN-amplified neuroblastoma. FOXR2 binds MYCN and appears to stabilize the MYCN protein, leading to high levels of MYCN protein in neuroblastoma with FOXR2 activation. This finding provides an explanation for the similar gene expression profiles for neuroblastoma with FOXR2 activation and neuroblastoma with MYCN amplification.
Neuroblastoma with FOXR2 activation is observed at comparable rates in high-risk and non–high-risk cases. Among high-risk cases, outcomes for patients whose tumors showed FOXR2 activation were similar to those for cases with MYCN amplification. In a multivariable analysis, FOXR2 activation was significantly associated with inferior OS, along with INSS stage 4, age 18 months or older, and MYCN amplification.
Exonic variants in neuroblastoma (including ALK variants and amplification)
Compared with adult cancers, pediatric neuroblastoma tumors show a low number of variants per genome that affect protein sequence (10–20 per genome). The most common gene variant is ALK, which is altered in approximately 10% of patients (see below). Other genes with even lower frequencies of variants include ATRX, PTPN11, ARID1A, and ARID1B. As shown in Figure 9, most neuroblastoma cases lack variants in genes that are altered in a recurrent manner.
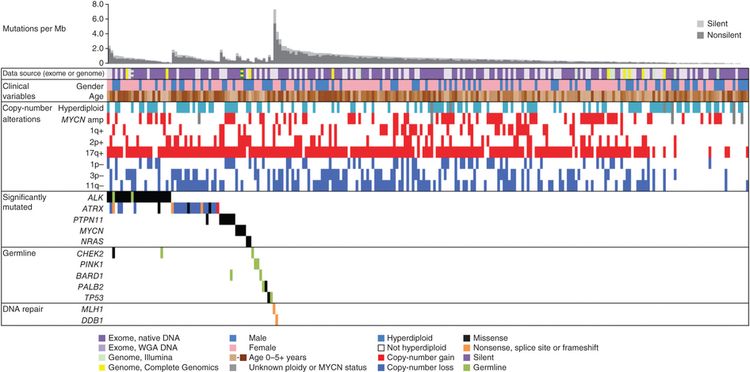 Figure 9. Data tracks (rows) facilitate the comparison of clinical and genomic data across cases with neuroblastoma (columns). The data sources and sequencing technology used were whole-exome sequencing (WES) from whole-genome amplification (WGA) (light purple), WES from native DNA (dark purple), Illumina WGS (green), and Complete Genomics WGS (yellow). Striped blocks indicate cases analyzed using two approaches. The clinical variables included were sex (male, blue; female, pink) and age (brown spectrum). Copy number alterations indicates ploidy measured by flow cytometry (with hyperdiploid meaning DNA index >1) and clinically relevant copy number alterations derived from sequence data. Significantly mutated genes are those with statistically significant mutation counts given the background mutation rate, gene size, and expression in neuroblastoma. Germline indicates genes with significant numbers of germline ClinVar variants or loss-of-function cancer gene variants in our cohort. DNA repair indicates genes that may be associated with an increased mutation frequency in two apparently hypermutated tumors. Predicted effects of somatic mutations are color coded according to the legend. Reprinted by permission from Macmillan Publishers Ltd: Nature Genetics (Pugh TJ, Morozova O, Attiyeh EF, et al.: The genetic landscape of high-risk neuroblastoma. Nat Genet 45 (3): 279-84, 2013), copyright (2013).
Figure 9. Data tracks (rows) facilitate the comparison of clinical and genomic data across cases with neuroblastoma (columns). The data sources and sequencing technology used were whole-exome sequencing (WES) from whole-genome amplification (WGA) (light purple), WES from native DNA (dark purple), Illumina WGS (green), and Complete Genomics WGS (yellow). Striped blocks indicate cases analyzed using two approaches. The clinical variables included were sex (male, blue; female, pink) and age (brown spectrum). Copy number alterations indicates ploidy measured by flow cytometry (with hyperdiploid meaning DNA index >1) and clinically relevant copy number alterations derived from sequence data. Significantly mutated genes are those with statistically significant mutation counts given the background mutation rate, gene size, and expression in neuroblastoma. Germline indicates genes with significant numbers of germline ClinVar variants or loss-of-function cancer gene variants in our cohort. DNA repair indicates genes that may be associated with an increased mutation frequency in two apparently hypermutated tumors. Predicted effects of somatic mutations are color coded according to the legend. Reprinted by permission from Macmillan Publishers Ltd: Nature Genetics (Pugh TJ, Morozova O, Attiyeh EF, et al.: The genetic landscape of high-risk neuroblastoma. Nat Genet 45 (3): 279-84, 2013), copyright (2013).
The ALK gene provides instructions for making a cell surface receptor tyrosine kinase, expressed at significant levels only in developing embryonic and neonatal brains. ALK is the exonic variant found most commonly in neuroblastoma. Germline variants in ALK have been identified as the major cause of hereditary neuroblastoma. Somatically acquired ALK-activating exonic are also found as oncogenic drivers in neuroblastoma.
Two large cohort studies examined the clinical correlates and prognostic significance of ALK alterations. One study from the COG examined ALK status in 1,596 diagnostic neuroblastoma samples across all risk groups. Another study from SIOPEN evaluated 1,092 patients with high-risk neuroblastoma.
- ALK tyrosine kinase domain variants occurred primarily at three hot spots (F1174, R1275, and F1245 positions), with 10% to 15% of variants occurring at other kinase domain positions.
- In the COG cohort, the frequency of ALK variants was 10% in the high-risk neuroblastoma group, 8% in the intermediate-risk neuroblastoma group, and 6% in the low-risk neuroblastoma group.
- In the SIOPEN high-risk population, ALK variants were divided into clonal (>20% variant allele frequency [VAF]) and subclonal (0.1%–20% VAF). Clonal ALK variants were detected in 10% of cases, and subclonal variants were found in 3.9% of patients. A total of 13.9% of the cases had an ALK variant.
- ALK variants were found at higher rates in patients with MYCN-amplified tumors compared with those without MYCN amplification: 10.9% versus 7.2%, respectively, for the COG cohort and 14% versus 6.5%, respectively, for the SIOPEN cohort (for clonal ALK variants).
- For patients with high-risk neuroblastoma, the ALK amplification was observed in approximately 4% of cases in both the COG and the SIOPEN cohorts. ALK amplification occurred almost exclusively in cases that also had MYCN amplification.
- ALK alterations were associated with inferior prognoses for high-risk neuroblastoma patients in both the COG and the SIOPEN studies:
- In the SIOPEN cohort, a statistically significant difference in OS was observed between cases with ALK amplification (ALKa) or clonal ALK variant (ALKm) versus subclonal ALKm or no ALK alterations (5-year OS rate: ALKa, 26% [95% CI, 10%–47%]; clonal ALKm, 33% [95% CI, 21%–44%]; subclonal ALKm, 48% [95% CI, 26%–67%]; and no alteration, 51% [95% CI, 46%–55%], respectively; P = .001). In a multivariate model, ALK amplification (HR, 2.38; P = .004) and clonal ALK variant (HR, 1.77; P = .001) were independent predictors of poor outcome.
- In the COG high-risk neuroblastoma population, inferior prognoses, similar to those seen in the SIOPEN cohort, were observed for cases with ALK variants and ALK amplifications.
In a study that compared the genomic data of primary diagnostic neuroblastomas originating in the adrenal gland (n = 646) with that of neuroblastomas originating in the thoracic sympathetic ganglia (n = 118), 16% of thoracic tumors harbored ALK variants.
Small-molecule ALK kinase inhibitors such as lorlatinib (added to conventional therapy) are being tested in patients with recurrent ALK-altered neuroblastoma (NCT03107988) and in patients with newly diagnosed high-risk neuroblastoma with activated ALK (COG ANBL1531). For more information, see the sections on Treatment of High-Risk Neuroblastoma and Treatment of Recurrent Neuroblastoma in Neuroblastoma Treatment.
Genomic evolution of exonic variants
There are limited data regarding the genomic evolution of exonic variants from diagnosis to relapse for neuroblastoma. Whole-genome sequencing was applied to 23 paired diagnostic and relapsed neuroblastoma tumor samples to define somatic genetic alterations associated with relapse, while a second study evaluated 16 paired diagnostic and relapsed specimens. Both studies identified an increased number of variants in the relapsed samples compared with the samples at diagnosis. This has been confirmed in a study of neuroblastoma tumor samples sent for next-generation sequencing.
- In the first study, an increased incidence of variants in genes associated with RAS-MAPK signaling was found in tumors at relapse compared with tumors from the same patient at diagnosis; 15 of 23 relapse samples contained somatic variants in genes involved in this pathway, and each variant was consistent with pathway activation.
In addition, three relapse samples showed structural alterations involving MAPK pathway genes consistent with pathway activation, so aberrations in this pathway were detected in 18 of 23 (78%) relapse samples. Aberrations were found in ALK (n = 10), NF1 (n = 2), and one each in NRAS, KRAS, HRAS, BRAF, PTPN11, and FGFR1. Even with deep sequencing, 7 of the 18 alterations were not detectable in the primary tumor, highlighting the evolution of variants presumably leading to relapse and the importance of genomic evaluations of tissues obtained at relapse.
- In the second study, ALK variants were not observed in either diagnostic or relapse specimens, but relapse-specific recurrent single-nucleotide variants were observed in 11 genes, including the putative CHD5 neuroblastoma tumor suppressor gene located at chromosome 1p36.
- A third retrospective variant-sequencing study used data from Foundation Medicine to compare tumor samples from patients with newly diagnosed neuroblastoma with tumor samples from patients with refractory and relapsed neuroblastoma. The study found a higher percentage of variants that were targetable with current drugs in the relapsed and refractory group.
- A fourth study evaluated the frequency of ALK alterations at diagnosis and relapse. There were significantly higher rates of ALK variants at relapse than at diagnosis (17.7% at relapse vs. 10.5% at diagnosis). The rate of ALK amplifications did not differ between diagnosis and relapse.
Given the widespread metastatic nature of high-risk and relapsed neuroblastoma, use of circulating tumor DNA (ctDNA) technologies may reveal additional genomic alterations not found in conventional tumor biopsies. Moreover, these approaches have demonstrated the ability to detect resistant variants in patients with neuroblastoma who were treated with ALK inhibitors.[Level of evidence C1] In one analysis of serial ctDNA samples from patients treated with lorlatinib, ALK variant allele frequency tracked with disease burden in most but not all patients. In subsets of patients who progressed while taking lorlatinib, second compound variants in ALK or variants in other genes, including RAS pathway genes, were reported.
In a deep-sequencing study, 276 neuroblastoma samples (comprised of all stages and from patients of all ages at diagnosis) underwent very deep (33,000X) sequencing of just two amplified ALK variant hot spots, which revealed 4.8% clonal variants and an additional 5% subclonal variants. This finding suggests that subclonal ALK gene variants are common. Thus, deep sequencing can reveal the presence of variants in tiny subsets of neuroblastoma tumor cells that may be able to survive during treatment and grow to constitute a relapse.
Genomic alterations promoting telomere maintenance
Lengthening of telomeres, the tips of chromosomes, promotes cell survival. Telomeres otherwise shorten with each cell replication, eventually resulting in the cell’s inability to replicate. Patients whose tumors lack telomere maintenance mechanisms have an excellent prognosis, while patients whose tumors harbored telomere maintenance mechanisms have a substantially worse prognosis. Low-risk neuroblastoma tumors, as defined by clinical/biological features, have little telomere lengthening activity. Aberrant genetic mechanisms for telomere lengthening have been identified in high-risk neuroblastoma tumors. Thus far, the following three mechanisms, which appear to be mutually exclusive, have been described:
- Chromosomal rearrangements involving a chromosomal region at 5p15.33 proximal to the TERT gene, which encodes the catalytic unit of telomerase, occur in approximately 20% to 25% of high-risk neuroblastoma cases and are mutually exclusive with MYCN amplifications and alternative lengthening of telomeres (ALT) activation. The rearrangements induce transcriptional upregulation of TERT by juxtaposing the TERT coding sequence with strong enhancer elements. Children whose tumors have TERT rearrangements have a poor prognosis, which is comparable to the prognosis of children whose tumors have MYCN amplification. Next-generation sequencing or fluorescence in situ hybridization (FISH) may be used to identify these alterations. One study identified TERT rearrangements by FISH in 6% of all patients with neuroblastic tumors regardless of risk group and in 12.4% of patients with high-risk neuroblastic tumors.
- Another mechanism promoting TERT overexpression is MYCN amplification, which is associated with approximately 40% to 50% of high-risk neuroblastoma cases.
- ALT is an additional mechanism of telomere maintenance that is used by neuroblastoma tumors. ALT activation is present in approximately 20% to 25% of newly diagnosed high-risk cases, compared with approximately 5% to 12% of low-risk and intermediate-risk cases. Compared with newly diagnosed cases, the proportion of neuroblastoma cases with ALT-positive tumors was higher in a cohort of patients who relapsed (10% vs. 48%, respectively). This finding may reflect the relatively indolent course of tumors with ALT activation after relapse, compared with the clinical course of other tumors after relapse. Over time, the proportion of patients with relapsed ALT-positive neuroblastomas (out of patients with neuroblastoma) appears larger than that of patients with another tumor type who relapsed (out of patients diagnosed with that tumor). Neuroblastoma cases with ALT activation have low TERT expression and can be identified by immunohistochemistry for the ALT-associated promyelocytic nuclear body, by FISH with a telomere probe to visualize telomere ultrabright spots, and by the C-circle assay. Approximately 55% to 60% of ALT-positive cases are characterized by deleterious ATRX variants. Cases lacking ATRX variants often show low ATRX protein expression.
ALT-positive tumors in pediatric populations rarely present before the age of 18 months and occur almost exclusively in older children (median age at diagnosis, approximately 8 years). The proportion of neuroblastoma cases with ATRX variants increases with age into the adolescent and young adult populations.
The prognosis for high-risk patients with ALT activation is as poor as that for patients with MYCN amplification for EFS; however, OS is more favorable for patients with ALT activation. The more favorable OS appears to result from a more protracted disease course after relapse, but with long-term survival at 10 to 15 years being as low as that for other high-risk neuroblastoma patients. In one report, EFS and OS for low-risk and intermediate-risk patients with ALT activation were similar to those observed for ALT-positive patients with high-risk disease.
Additional biological factors associated with prognosis
MYC and MYCN expression
Immunostaining for MYC and MYCN proteins on a restricted subset of 357 undifferentiated/poorly differentiated neuroblastoma tumors demonstrated that elevated MYC/MYCN protein expression is prognostically significant. Sixty-eight tumors (19%) highly expressed the MYCN protein, and 81 were MYCN amplified. Thirty-nine tumors (10.9%) expressed MYC highly and were mutually exclusive of high MYCN expression. In the MYC-expressing tumors, MYC or MYCN gene amplification was not seen. Segmental chromosomal aberrations were not examined in this study.
- Patients with favorable-histology tumors without high MYC/MYCN expression had favorable survival (3-year EFS rate, 89.7% ± 5.5%; 3-year OS rate, 97% ± 3.2%).
- Patients with undifferentiated or poorly differentiated histology tumors without MYC/MYCN expression had a 3-year EFS rate of 63.1% (± 13.6%) and a 3-year OS rate of 83.5% (± 9.4%).
- Three-year EFS rates in patients with MYCN amplification, high MYCN expression, and high MYC expression were 48.1% (± 11.5%), 46.2% (± 12%), and 43.4% (± 23.1%), respectively. OS rates were 65.8% (± 11.1%), 63.2% (± 12.1%), and 63.5% (± 19.2%), respectively.
- Additionally, when high expression of MYC and MYCN proteins underwent multivariate analysis with other prognostic factors, including MYC/MYCN gene amplification, high MYC and MYCN protein expression was independent of other prognostic markers.
Neurotrophin receptor kinases
Expression of neurotrophin receptor kinases and their ligands vary between high-risk and low-risk tumors. TrkA is found on low-risk tumors, and absence of its ligand NGF is postulated to lead to spontaneous tumor regression. In contrast, TrkB is found in high-risk tumors that also express its ligand, BDNF, which promotes neuroblastoma cell growth and survival.
For information about the treatment of neuroblastoma, see Neuroblastoma Treatment.
References
- Cohn SL, Pearson AD, London WB, et al.: The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 27 (2): 289-97, 2009.
- Schleiermacher G, Mosseri V, London WB, et al.: Segmental chromosomal alterations have prognostic impact in neuroblastoma: a report from the INRG project. Br J Cancer 107 (8): 1418-22, 2012.
- Janoueix-Lerosey I, Schleiermacher G, Michels E, et al.: Overall genomic pattern is a predictor of outcome in neuroblastoma. J Clin Oncol 27 (7): 1026-33, 2009.
- Schleiermacher G, Michon J, Ribeiro A, et al.: Segmental chromosomal alterations lead to a higher risk of relapse in infants with MYCN-non-amplified localised unresectable/disseminated neuroblastoma (a SIOPEN collaborative study). Br J Cancer 105 (12): 1940-8, 2011.
- Carén H, Kryh H, Nethander M, et al.: High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc Natl Acad Sci U S A 107 (9): 4323-8, 2010.
- Schleiermacher G, Janoueix-Lerosey I, Ribeiro A, et al.: Accumulation of segmental alterations determines progression in neuroblastoma. J Clin Oncol 28 (19): 3122-30, 2010.
- Defferrari R, Mazzocco K, Ambros IM, et al.: Influence of segmental chromosome abnormalities on survival in children over the age of 12 months with unresectable localised peripheral neuroblastic tumours without MYCN amplification. Br J Cancer 112 (2): 290-5, 2015.
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.
- Ambros IM, Tonini GP, Pötschger U, et al.: Age Dependency of the Prognostic Impact of Tumor Genomics in Localized Resectable MYCN-Nonamplified Neuroblastomas. Report From the SIOPEN Biology Group on the LNESG Trials and a COG Validation Group. J Clin Oncol 38 (31): 3685-3697, 2020.
- Pinto N, Naranjo A, Ding X, et al.: Impact of Genomic and Clinical Factors on Outcome of Children ≥18 Months of Age with Stage 3 Neuroblastoma with Unfavorable Histology and without MYCN Amplification: A Children's Oncology Group (COG) Report. Clin Cancer Res 29 (8): 1546-1556, 2023.
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.
- Pinto N, Naranjo A, Hibbitts E, et al.: Predictors of differential response to induction therapy in high-risk neuroblastoma: A report from the Children's Oncology Group (COG). Eur J Cancer 112: 66-79, 2019.
- Depuydt P, Boeva V, Hocking TD, et al.: Genomic Amplifications and Distal 6q Loss: Novel Markers for Poor Survival in High-risk Neuroblastoma Patients. J Natl Cancer Inst 110 (10): 1084-1093, 2018.
- Ambros PF, Ambros IM, Brodeur GM, et al.: International consensus for neuroblastoma molecular diagnostics: report from the International Neuroblastoma Risk Group (INRG) Biology Committee. Br J Cancer 100 (9): 1471-82, 2009.
- Kreissman SG, Seeger RC, Matthay KK, et al.: Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): a randomised phase 3 trial. Lancet Oncol 14 (10): 999-1008, 2013.
- Bagatell R, Beck-Popovic M, London WB, et al.: Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol 27 (3): 365-70, 2009.
- Plantaz D, Vandesompele J, Van Roy N, et al.: Comparative genomic hybridization (CGH) analysis of stage 4 neuroblastoma reveals high frequency of 11q deletion in tumors lacking MYCN amplification. Int J Cancer 91 (5): 680-6, 2001.
- Maris JM, Hogarty MD, Bagatell R, et al.: Neuroblastoma. Lancet 369 (9579): 2106-20, 2007.
- Campbell K, Shyr D, Bagatell R, et al.: Comprehensive evaluation of context dependence of the prognostic impact of MYCN amplification in neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer 66 (8): e27819, 2019.
- Berbegall AP, Bogen D, Pötschger U, et al.: Heterogeneous MYCN amplification in neuroblastoma: a SIOP Europe Neuroblastoma Study. Br J Cancer 118 (11): 1502-1512, 2018.
- Schmitt-Hoffner F, van Rijn S, Toprak UH, et al.: FOXR2 Stabilizes MYCN Protein and Identifies Non-MYCN-Amplified Neuroblastoma Patients With Unfavorable Outcome. J Clin Oncol 39 (29): 3217-3228, 2021.
- Sturm D, Orr BA, Toprak UH, et al.: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5): 1060-72, 2016.
- Pugh TJ, Morozova O, Attiyeh EF, et al.: The genetic landscape of high-risk neuroblastoma. Nat Genet 45 (3): 279-84, 2013.
- Peifer M, Hertwig F, Roels F, et al.: Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526 (7575): 700-4, 2015.
- Valentijn LJ, Koster J, Zwijnenburg DA, et al.: TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet 47 (12): 1411-4, 2015.
- Cheung NK, Zhang J, Lu C, et al.: Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 307 (10): 1062-71, 2012.
- Molenaar JJ, Koster J, Zwijnenburg DA, et al.: Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483 (7391): 589-93, 2012.
- Sausen M, Leary RJ, Jones S, et al.: Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet 45 (1): 12-7, 2013.
- Bresler SC, Weiser DA, Huwe PJ, et al.: ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 26 (5): 682-94, 2014.
- Janoueix-Lerosey I, Lequin D, Brugières L, et al.: Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455 (7215): 967-70, 2008.
- Bellini A, Pötschger U, Bernard V, et al.: Frequency and Prognostic Impact of ALK Amplifications and Mutations in the European Neuroblastoma Study Group (SIOPEN) High-Risk Neuroblastoma Trial (HR-NBL1). J Clin Oncol 39 (30): 3377-3390, 2021.
- Oldridge DA, Truong B, Russ D, et al.: Differences in Genomic Profiles and Outcomes Between Thoracic and Adrenal Neuroblastoma. J Natl Cancer Inst 111 (11): 1192-1201, 2019.
- Eleveld TF, Oldridge DA, Bernard V, et al.: Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 47 (8): 864-71, 2015.
- Schramm A, Köster J, Assenov Y, et al.: Mutational dynamics between primary and relapse neuroblastomas. Nat Genet 47 (8): 872-7, 2015.
- Padovan-Merhar OM, Raman P, Ostrovnaya I, et al.: Enrichment of Targetable Mutations in the Relapsed Neuroblastoma Genome. PLoS Genet 12 (12): e1006501, 2016.
- Rosswog C, Fassunke J, Ernst A, et al.: Genomic ALK alterations in primary and relapsed neuroblastoma. Br J Cancer 128 (8): 1559-1571, 2023.
- Bosse KR, Giudice AM, Lane MV, et al.: Serial Profiling of Circulating Tumor DNA Identifies Dynamic Evolution of Clinically Actionable Genomic Alterations in High-Risk Neuroblastoma. Cancer Discov 12 (12): 2800-2819, 2022.
- Berko ER, Witek GM, Matkar S, et al.: Circulating tumor DNA reveals mechanisms of lorlatinib resistance in patients with relapsed/refractory ALK-driven neuroblastoma. Nat Commun 14 (1): 2601, 2023.
- Bellini A, Bernard V, Leroy Q, et al.: Deep Sequencing Reveals Occurrence of Subclonal ALK Mutations in Neuroblastoma at Diagnosis. Clin Cancer Res 21 (21): 4913-21, 2015.
- Ackermann S, Cartolano M, Hero B, et al.: A mechanistic classification of clinical phenotypes in neuroblastoma. Science 362 (6419): 1165-1170, 2018.
- Roderwieser A, Sand F, Walter E, et al.: Telomerase is a prognostic marker of poor outcome and a therapeutic target in neuroblastoma. JCO Precis Oncol 3: 1-20, 2019.
- Yu Y, Zhang M, Yao X, et al.: Translational practice of fluorescence in situ hybridisation to identify neuroblastic tumours with TERT rearrangements. J Pathol Clin Res 9 (6): 475-487, 2023.
- Mac SM, D'Cunha CA, Farnham PJ: Direct recruitment of N-myc to target gene promoters. Mol Carcinog 29 (2): 76-86, 2000.
- Hartlieb SA, Sieverling L, Nadler-Holly M, et al.: Alternative lengthening of telomeres in childhood neuroblastoma from genome to proteome. Nat Commun 12 (1): 1269, 2021.
- Koneru B, Lopez G, Farooqi A, et al.: Telomere Maintenance Mechanisms Define Clinical Outcome in High-Risk Neuroblastoma. Cancer Res 80 (12): 2663-2675, 2020.
- Wang LL, Teshiba R, Ikegaki N, et al.: Augmented expression of MYC and/or MYCN protein defines highly aggressive MYC-driven neuroblastoma: a Children's Oncology Group study. Br J Cancer 113 (1): 57-63, 2015.
- Maris JM, Matthay KK: Molecular biology of neuroblastoma. J Clin Oncol 17 (7): 2264-79, 1999.
Retinoblastoma
Genomics of Retinoblastoma
Retinoblastoma is a tumor that occurs in heritable (25%–30%) and nonheritable (70%–75%) forms. Heritable disease is defined by the presence of a germline variant of the RB1 gene. This germline variant may have been inherited from an affected progenitor (25% of cases) or may have occurred in a germ cell before conception or in utero during early embryogenesis in patients with sporadic disease (75% of cases). The presence of positive family history or bilateral or multifocal disease is suggestive of heritable disease.
Heritable retinoblastoma may manifest as unilateral or bilateral disease. The penetrance of the RB1 variant (laterality, age at diagnosis, and number of tumors) is probably dependent on concurrent genetic modifiers such as MDM2 and MDM4 polymorphisms. All children with bilateral disease and approximately 15% of patients with unilateral disease are presumed to have the heritable form, even though only 25% have an affected parent. In a series of 482 patients with unilateral retinoblastoma, germline variants were identified in 33% of infants younger than 12 months, 6% of children aged 12 to 24 months, and 7% of children aged 24 to 39 months. The highest incidence of germline retinoblastoma was in patients younger than 1 year compared with patients older than 1 year (odds ratio, 2.96).[Level of evidence C2]
Children with heritable retinoblastoma tend to be diagnosed at a younger age than are children with the nonheritable form of the disease.
The genomic landscape of retinoblastoma is driven by alterations in RB1 that lead to biallelic inactivation. A rare cause of RB1 inactivation is chromothripsis, which may be difficult to detect by conventional methods.
Recurrent changes in genes other than RB1 are uncommon in retinoblastoma but do occur. Variants or deletions of BCOR and amplification of MYCN are the most frequently reported events. A study of 1,068 unilateral nonfamilial retinoblastoma tumors reported that 2% to 3% of tumors lacked evidence of RB1 loss and approximately one-half of these cases without evidence of RB1 loss showed MYCN amplification. However, MYCN amplification is also observed in retinoblastoma tumors that have RB1 alterations, suggesting that inactivation of RB1 by a variant or an inactive retinoblastoma protein is a requirement for the development of retinoblastoma, independent of MYCN amplification.
Genetic counseling is recommended for all patients with retinoblastoma. For more information, see the Genetic Counseling section.
For information about the treatment of retinoblastoma, see Retinoblastoma Treatment.
References
- Castéra L, Sabbagh A, Dehainault C, et al.: MDM2 as a modifier gene in retinoblastoma. J Natl Cancer Inst 102 (23): 1805-8, 2010.
- de Oliveira Reis AH, de Carvalho IN, de Sousa Damasceno PB, et al.: Influence of MDM2 and MDM4 on development and survival in hereditary retinoblastoma. Pediatr Blood Cancer 59 (1): 39-43, 2012.
- Shields CL, Dockery P, Ruben M, et al.: Likelihood of Germline Mutation With Solitary Unilateral Retinoblastoma Based on Patient Age at Presentation: Analysis of 482 Consecutive Patients. J Pediatr Ophthalmol Strabismus 58 (6): 355-364, 2021 Nov-Dec.
- Andreoli MT, Chau FY, Shapiro MJ, et al.: Epidemiological trends in 1452 cases of retinoblastoma from the Surveillance, Epidemiology, and End Results (SEER) registry. Can J Ophthalmol 52 (6): 592-598, 2017.
- Zhang J, Benavente CA, McEvoy J, et al.: A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 481 (7381): 329-34, 2012.
- Rushlow DE, Mol BM, Kennett JY, et al.: Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol 14 (4): 327-34, 2013.
- McEvoy J, Nagahawatte P, Finkelstein D, et al.: RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget 5 (2): 438-50, 2014.
- Afshar AR, Pekmezci M, Bloomer MM, et al.: Next-Generation Sequencing of Retinoblastoma Identifies Pathogenic Alterations beyond RB1 Inactivation That Correlate with Aggressive Histopathologic Features. Ophthalmology 127 (6): 804-813, 2020.
- Kooi IE, Mol BM, Massink MP, et al.: Somatic genomic alterations in retinoblastoma beyond RB1 are rare and limited to copy number changes. Sci Rep 6: 25264, 2016.
- Francis JH, Richards AL, Mandelker DL, et al.: Molecular Changes in Retinoblastoma beyond RB1: Findings from Next-Generation Sequencing. Cancers (Basel) 13 (1): , 2021.
- Ewens KG, Bhatti TR, Moran KA, et al.: Phosphorylation of pRb: mechanism for RB pathway inactivation in MYCN-amplified retinoblastoma. Cancer Med 6 (3): 619-630, 2017.
Kidney Tumors
Wilms Tumor
Molecular Features of Wilms Tumor
A Wilms tumor may arise during embryogenesis on the background of an otherwise genomically normal kidney, or it may arise from nongermline somatic genetic precursor lesions residing in histologically and functionally normal kidney tissue. Hypermethylation of H19, a known component of a subset of Wilms tumors, is a very common genetic abnormality found in these normal-appearing areas of precursor lesions.
One study performed genome-wide sequencing, mRNA and miRNA expression, DNA copy number, and methylation analysis on 117 Wilms tumors, followed by targeted sequencing of 651 Wilms tumors. The tumors were selected for either favorable histology (FH) Wilms that had relapsed or those with diffuse anaplasia. The study showed the following:
- Wilms tumors commonly arise through more than one genetic event.
- Wilms tumors show differences in gene expression and methylation patterns with different genetic aberrations.
- Wilms tumors have a large number of candidate driver genes, most of which are altered in less than 5% of Wilms tumors.
- Wilms tumors have recurrent variants in genes with common functions, with most involved in either early renal development or epigenetic regulation (e.g., chromatin modifications, transcription elongation, and miRNA).
Approximately one-third of Wilms tumor cases involve variants in WT1, CTNNB1, or AMER1 (WTX). Another subset of Wilms tumor cases results from variants in miRNA processing genes (miRNAPG), including DROSHA, DGCR8, DICER1, and XPO5. Other genes critical for early renal development that are recurrently altered in Wilms tumor include SIX1 and SIX2 (transcription factors that play key roles in early renal development), EP300, CREBBP, and MYCN. Of the variants in Wilms tumors, 30% to 50% appear to converge on the process of transcriptional elongation in renal development and include the genes MLLT1, BCOR, MAP3K4, BRD7, and HDAC4. Anaplastic Wilms tumor is characterized by the presence of TP53 variants.
Elevated rates of Wilms tumor are observed in patients with a number of genetic disorders, including WAGR (Wilms tumor, aniridia, genitourinary abnormalities, and range of developmental delays) syndrome (WAGR spectrum), Beckwith-Wiedemann syndrome, hemihypertrophy, Denys-Drash syndrome, and Perlman syndrome. Other genetic causes that have been observed in familial Wilms tumor cases include germline variants in REST and CTR9.
The genomic and genetic characteristics of Wilms tumor are summarized below.
WT1 gene
The WT1 gene is located on the short arm of chromosome 11 (11p13). WT1 is a transcription factor that is required for normal genitourinary development and is important for differentiation of the renal blastema. WT1 variants are observed in 10% to 20% of cases of sporadic Wilms tumor.
Wilms tumor with a WT1 variant is characterized by the following:
- Evidence of WNT pathway activation by activating variants in the CTNNB1 gene is common.
- Loss of heterozygosity (LOH) at 11p15 is commonly observed, as paternal uniparental disomy for chromosome 11 represents a common mechanism for losing the remaining normal WT1 allele.
- Nephrogenic rests are benign foci of embryonal kidney cells that abnormally persist into postnatal life. Intralobar nephrogenic rests occur in approximately 20% of Wilms tumor cases. They are observed at high rates in cases with genetic syndromes that have WT1 variants such as WAGR and Denys-Drash syndromes. Intralobar nephrogenic rests are also observed in cases with sporadic WT1 and MLLT1 variants.
- WT1 germline variants are uncommon (2%–4%) in nonsyndromic Wilms tumor.
- WT1 variants and 11p15 LOH were associated with relapse in patients with very low-risk Wilms tumor in one study of 56 patients who did not receive chemotherapy. These findings need validation but may provide biomarkers for stratifying patients in the future.
Germline WT1 variants are more common in children with Wilms tumor and one of the following:
- WAGR syndrome, Denys-Drash syndrome, or Frasier syndrome.
- Genitourinary anomalies, including hypospadias and cryptorchidism.
- Bilateral Wilms tumor.
- Unilateral Wilms tumor with nephrogenic rests in the contralateral kidney.
- Stromal and rhabdomyomatous differentiation.
Germline WT1 point variants produce genetic syndromes that are characterized by nephropathy, 46XY disorder of sex development, and varying risks of Wilms tumor. Syndromic conditions with germline WT1 variants include WAGR syndrome, Denys-Drash syndrome, and Frasier syndrome.
- WAGR syndrome. Children with WAGR syndrome are at high risk (approximately 50%) of developing Wilms tumor. WAGR syndrome results from deletions at chromosome 11p13 that involve a set of contiguous genes that include the WT1 and PAX6 genes.
Inactivating variants or deletions in the PAX6 gene lead to aniridia, while deletion of WT1 confers the increased risk of Wilms tumor. Loss of the LMO2 gene has been associated with a more frequent development of Wilms tumor in patients with congenital aniridia and WAGR-region deletions.[Level of evidence C1] Sporadic aniridia in which WT1 is not deleted is not associated with increased risk of Wilms tumor. Accordingly, children with familial aniridia, generally occurring for many generations, and without renal abnormalities, have a normal WT1 gene and are not at an increased risk of Wilms tumor.
Wilms tumor in children with WAGR syndrome is characterized by an excess of bilateral disease, intralobar nephrogenic rests, early age at diagnosis, and stromal-predominant histology in FH tumors. The intellectual disability in WAGR syndrome may be secondary to deletion of other genes, including SLC1A2 or BDNF.
- Denys-Drash syndrome. This syndrome is characterized by nephrotic syndrome caused by diffuse mesangial sclerosis, XY pseudohermaphroditism, and increased risk of Wilms tumor (>90%).
WT1 variants in Denys-Drash syndrome are most often missense variants in exons 8 and 9, which code for the DNA binding region of WT1.
- Frasier syndrome. This syndrome is characterized by progressive nephropathy caused by focal segmental glomerulosclerosis, gonadoblastoma, and XY pseudohermaphroditism.
WT1 variants in Frasier syndrome typically occur in intron 9 at the KT site, and create an alternative splicing variant, thereby preventing production of the usually more abundant WT1 +KTS isoform.
Studies evaluating genotype/phenotype correlations of WT1 variants have shown that the risk of Wilms tumor is highest for truncating variants (14 of 17 cases, 82%) and lower for missense variants (27 of 67 cases, 42%). The risk is lowest for KTS splice site variants (1 of 27 cases, 4%). Bilateral Wilms tumor was more common in cases with WT1-truncating variants (9 of 14 cases) than in cases with WT1 missense variants (3 of 27 cases). These genomic studies confirm previous estimates of elevated risk of Wilms tumor for children with Denys-Drash syndrome and low risk of Wilms tumor for children with Frasier syndrome.
CTNNB1 gene
CTNNB1 is one of the most commonly altered genes in Wilms tumor, reported to occur in 15% of patients with Wilms tumor. These CTNNB1 variants result in activation of the WNT pathway, which plays a prominent role in the developing kidney. CTNNB1 variants commonly occur with WT1 variants, and most cases of Wilms tumor with WT1 variants have a concurrent CTNNB1 variant. Activation of beta-catenin in the presence of intact WT1 protein appears to be inadequate to promote tumor development because CTNNB1 variants are rarely found in the absence of a WT1 or AMER1 variant, except when associated with a MLLT1 variant. CTNNB1 variants appear to be late events in Wilms tumor development because they are found in tumors but not in nephrogenic rests.
AMER1 (WTX) gene on the X chromosome
AMER1 is located on the X chromosome at Xq11.1. It is altered in 15% to 20% of Wilms tumor cases. Germline variants in AMER1 cause an X-linked sclerosing bone dysplasia, osteopathia striata congenita with cranial sclerosis (MIM300373). Despite having germline AMER1 variants, individuals with osteopathia striata congenita are not predisposed to tumor development. The AMER1 protein appears to be involved in both the degradation of beta-catenin and in the intracellular distribution of APC protein. AMER1 is most commonly altered by deletions involving part or all of the AMER1 gene, with deleterious point variants occurring less commonly. Most Wilms tumor cases with AMER1 alterations have epigenetic 11p15 abnormalities.
AMER1 alterations are equally distributed between males and females, and AMER1 inactivation has no apparent effect on clinical presentation or prognosis.
Imprinting cluster regions (ICRs) on chromosome 11p15 (WT2) and Beckwith-Wiedemann syndrome
A second Wilms tumor locus, WT2, maps to an imprinted region of chromosome 11p15.5. When it is a germline variant, it causes Beckwith-Wiedemann syndrome. About 3% of children with Wilms tumor have germline epigenetic or genetic changes at the 11p15.5 growth regulatory locus without any clinical manifestations of overgrowth. Like children with Beckwith-Wiedemann syndrome, these children have an increased incidence of bilateral Wilms tumor or familial Wilms tumor.
Approximately one-fifth of patients with Beckwith-Wiedemann syndrome who develop Wilms tumor present with bilateral disease, and metachronous bilateral disease is also observed. The prevalence of Beckwith-Wiedemann syndrome is about 1% among children with Wilms tumor reported to the National Wilms Tumor Study (NWTS).
Approximately 80% of patients with Beckwith-Wiedemann syndrome have a molecular defect of the 11p15 domain. Various molecular mechanisms underlying Beckwith-Wiedemann syndrome have been identified. Some of these abnormalities are genetic (germline variants of the maternal allele of CDKN1C, paternal uniparental isodisomy of 11p15, or duplication of part of the 11p15 domain) but are more frequently epigenetic (loss of methylation of the maternal ICR2 [CDKN1C and KCNQ1OT1 genes] or gain of methylation of the maternal ICR1 [IGF2 and H19 genes]).
Several candidate genes at the WT2 locus comprise the two independent imprinted domains: IGF2 and H19; and CDKN1C and KCNQ1OT1. LOH, which exclusively affects the maternal chromosome, has the effect of upregulating paternally active genes and silencing maternally active ones. A loss or switch of the imprint for genes (change in methylation status) in this region has also been frequently observed and results in the same functional aberrations.
A relationship between epigenotype and phenotype has been shown in Beckwith-Wiedemann syndrome, with a different rate of cancer in Beckwith-Wiedemann syndrome according to the type of alteration of the 11p15 region.
The following four main molecular subtypes of Beckwith-Wiedemann syndrome are characterized by specific genotype-phenotype correlations:
- ICR1 gain of methylation (ICR1-GoM). Five percent to 10% of cases are caused by telomeric ICR1-GoM, which causes both biallelic expression of the IGF2 gene (normally expressed by the paternal allele only) and reduced expression of the oncosuppressor H19 gene. The incidence of Wilms tumor is 22.8%.
- ICR2 loss of methylation (ICR2-LoM). Fifty percent of cases with Beckwith-Wiedemann syndrome are caused by ICR2-LoM, resulting in reduced expression of the CDKN1C gene, normally expressed by the maternal chromosome only. Tumor incidence is very low (2.5%).
- Uniparental disomy (UPD). Altered expression at both imprinted gene clusters is observed in mosaic UPD of chromosome 11p15.5, accounting for 20% to 25% of the cases. The incidence of Wilms tumor is 6.2%, followed by hepatoblastoma (4.7%) and adrenal carcinoma (1.5%). Fewer than 1% of cases with Beckwith-Wiedemann syndrome are caused by chromosomal rearrangements involving the 11p15 region.
- CDKN1C variants. Maternally inheritable CDKN1C loss-of-function variants account for approximately 5% of the cases. This type is associated with a 4.3% incidence of neuroblastoma.
Other tumors such as neuroblastoma or hepatoblastoma were reported in patients with paternal 11p15 isodisomy. For patients with Beckwith-Wiedemann syndrome, the relative risk of developing hepatoblastoma is 2,280 times that of the general population.
Loss of imprinting or gene methylation is rarely found at other loci, supporting the specificity of loss of imprinting at 11p15.5. Interestingly, Wilms tumor in Japanese and East Asian children, which occurs at a lower incidence than in White children, is not associated with either nephrogenic rests or IGF2 loss of imprinting.
Other genes and chromosomal alterations
Additional genes and chromosomal alterations that have been implicated in the pathogenesis and biology of Wilms tumor include the following:
- 1q. Gain of chromosome 1q is associated with an inferior outcome and is the single most powerful predictor of outcome. Gain of chromosome 1q is one of the most common cytogenetic abnormalities in Wilms tumor and is observed in approximately 30% of tumors.
In an analysis of FH Wilms tumor from 1,114 patients from NWTS-5 (COG-Q9401/NCT00002611), 28% of the tumors displayed 1q gain.
- The 8-year event-free survival (EFS) rate was 77% for patients with 1q gain and 90% for those lacking 1q gain (P< .001). Within each disease stage, 1q gain was associated with inferior EFS.
- The 8-year overall survival (OS) rate was 88% for those with 1q gain and 96% for those lacking 1q gain (P< .001). OS was significantly inferior in cases with stage I disease (P< .0015) and stage IV disease (P = .011).
- Similar results were reported in the International Society of Paediatric Oncology (SIOP) WT 2001 study of 586 children with Wilms tumor.
One study included a cohort of FH Wilms tumor that was enriched for patients who relapsed. The study found that the prevalence of 1q gain was higher in the relapsed Wilms tumor specimens (75%) than in the matched primary samples (47%). The increased prevalence of 1q gain at relapse supports its association with poor prognosis and disease progression.
- 16q and 1p. Additional tumor-suppressor or tumor-progression genes may lie on chromosomes 16q and 1p, as evidenced by LOH for these regions in 17% and 11% of Wilms tumor cases, respectively.
- In large NWTS studies, patients with tumor-specific loss of these loci had significantly worse relapse-free survival and OS rates. Combined loss of 1p and 16q are criteria used to select FH Wilms tumor patients for more aggressive therapy in the current Children's Oncology Group (COG) study. However, a U.K. study of more than 400 patients found no significant association between 1p deletion and poor prognosis, but a poor prognosis was associated with 16q LOH.
- An Italian study of 125 patients, using treatment quite similar to that in the COG study, found significantly worse prognosis in those with 1p deletions but not 16q deletions.
These conflicting results may arise from the greater prognostic significance of 1q gain described above. LOH of 16q and 1p loses significance as independent prognostic markers in the presence of 1q gain. However, in the absence of 1q gain, LOH of 16q and 1p retains their adverse prognostic impact. The LOH of 16q and 1p appears to arise from complex chromosomal events that result in 1q LOH or 1q gain. The change in 1q appears to be the critical tumorigenic genetic event.
- miRNAPG. Variants in selected miRNAPG are observed in approximately 20% of Wilms tumor cases and appear to perpetuate the progenitor state. The products of these genes direct the maturation of miRNAs from the initial pre-miRNA transcripts to functional cytoplasmic miRNAs (see Figure 10). The most commonly altered miRNAPG is DROSHA, with a recurrent variant (E1147K) affecting a metal-binding residue of the RNase IIIb domain, representing about 80% of DROSHA-altered tumors. Other miRNAPG that are altered in Wilms tumor include DGCR8, DICER1, TARBP2, DIS3L2, and XPO5. These variants are generally mutually exclusive, and they appear to be deleterious and result in impaired expression of tumor-suppressing miRNAs. A striking sex bias was noted for patients with variants in DGCR8 (located on chromosome 22q11), with 38 of 43 cases (88%) arising in girls.
Germline variants in miRNAPG are observed for DICER1 and DIS3L2, with variants in the former causing DICER1 syndrome and variants in the latter causing Perlman syndrome.
- DICER1 syndrome is typically caused by inherited truncating variants in DICER1, with tumor formation following acquisition of a missense variant in a domain of the remaining allele of DICER1 (the RNase IIIb domain) responsible for processing miRNAs derived from the 5p arms of pre-miRNAs. Tumors associated with DICER1 syndrome include pleuropulmonary blastoma, cystic nephroma, ovarian sex cord–stromal tumors, multinodular goiter, and embryonal rhabdomyosarcoma. Wilms tumor is an uncommon presentation of the DICER1 syndrome. In one study, three families with DICER1 syndrome included children with Wilms tumor, with two of the Wilms tumor cases showing the typical second DICER1 variant in the RNase IIIb domain. Another study identified DICER1 variants in 2 of 48 familial Wilms tumor families. Large sequencing studies of Wilms tumor cohorts have also observed occasional cases with DICER1 variants.
- Perlman syndrome is a rare autosomal recessive overgrowth disorder caused by variants in DIS3L2, which encodes a ribonuclease that is responsible for degrading pre-let-7 miRNA. Heterozygous DIS3L2 germline inactivations are also associated with Wilms tumor development. Patients with Perlman syndrome have a poor prognosis, with a high neonatal mortality rate. In a survey of published cases of Perlman syndrome (N = 28), in infants who survived beyond the neonatal period, approximately two-thirds developed Wilms tumor, and all patients showed developmental delay. Fetal macrosomia, ascites, and polyhydramnios are frequent manifestations.
 Figure 10. The miRNA processing pathway is commonly mutated in Wilms tumor. Expression of mature miRNA is initiated by RNA polymerase–mediated transcription of DNA-encoded sequences into pri-miRNA, which form a long double-stranded hairpin. This structure is then cleaved by a complex of Drosha and DGCR8 into a smaller pre-miRNA hairpin, which is exported from the nucleus and then cleaved by Dicer (an RNase) and TRBP (with specificity for dsRNA) to remove the hairpin loop and leave two single-stranded miRNAs. The functional strand binds to Argonaute (Ago2) proteins into the RNA-induced silencing complex (RISC), where it guides the complex to its target mRNA, while the nonfunctional strand is degraded. Targeting of mRNAs by this method results in mRNA silencing by mRNA cleavage, translational repression, or deadenylation. Let-7 miRNAs are a family of miRNAs highly expressed in ESCs with tumor suppressor properties. In cases in which LIN28 is overexpressed, LIN28 binds to pre-Let-7 miRNA, preventing DICER from binding and resulting in LIN28-activated polyuridylation by TUT4 or TUT7, causing reciprocal DIS3L2-mediated degradation of Let-7 pre-miRNAs. Genes involved in miRNA processing that have been associated with Wilms tumor are highlighted in blue (inactivating) and green (activating) and include DROSHA, DGCR8, XPO5 (encoding exportin-5), DICER1, TARBP2, DIS3L2, and LIN28. Copyright © 2015 Hohenstein et al.; Published by Cold Spring Harbor Laboratory Press. Genes Dev. 2015 Mar 1; 29(5): 467–482. doi: 10.1101/gad.256396.114. This article is distributed exclusively by Cold Spring Harbor Laboratory Press under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
Figure 10. The miRNA processing pathway is commonly mutated in Wilms tumor. Expression of mature miRNA is initiated by RNA polymerase–mediated transcription of DNA-encoded sequences into pri-miRNA, which form a long double-stranded hairpin. This structure is then cleaved by a complex of Drosha and DGCR8 into a smaller pre-miRNA hairpin, which is exported from the nucleus and then cleaved by Dicer (an RNase) and TRBP (with specificity for dsRNA) to remove the hairpin loop and leave two single-stranded miRNAs. The functional strand binds to Argonaute (Ago2) proteins into the RNA-induced silencing complex (RISC), where it guides the complex to its target mRNA, while the nonfunctional strand is degraded. Targeting of mRNAs by this method results in mRNA silencing by mRNA cleavage, translational repression, or deadenylation. Let-7 miRNAs are a family of miRNAs highly expressed in ESCs with tumor suppressor properties. In cases in which LIN28 is overexpressed, LIN28 binds to pre-Let-7 miRNA, preventing DICER from binding and resulting in LIN28-activated polyuridylation by TUT4 or TUT7, causing reciprocal DIS3L2-mediated degradation of Let-7 pre-miRNAs. Genes involved in miRNA processing that have been associated with Wilms tumor are highlighted in blue (inactivating) and green (activating) and include DROSHA, DGCR8, XPO5 (encoding exportin-5), DICER1, TARBP2, DIS3L2, and LIN28. Copyright © 2015 Hohenstein et al.; Published by Cold Spring Harbor Laboratory Press. Genes Dev. 2015 Mar 1; 29(5): 467–482. doi: 10.1101/gad.256396.114. This article is distributed exclusively by Cold Spring Harbor Laboratory Press under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
- SIX1 and SIX2.SIX1 and SIX2 are highly homologous transcription factors that play key roles in early renal development and are expressed in the metanephric mesenchyme, where they maintain the mesenchymal progenitor population. In patients with Wilms tumors, the frequency of SIX1 variants is 3% to 4%, and the frequency of SIX2 variants is 1% to 3%.
- Virtually all SIX1 and SIX2 variants are in exon 1 and result in a glutamine-to-arginine variant at position 177 (Q177R).
- Variants in WT1, AMER1, and CTNNB1 are infrequent in cases with SIX1, SIX2, or miRNAPG variants. Conversely, SIX1 or SIX2 variants and miRNAPG variants tend to occur together.
- In Wilms tumor, SIX1 and SIX2 variants are associated with the high-risk blastemal subtype and the presence of undifferentiated blastema in chemotherapy-naïve samples.
- In a study of 82 cases of FH Wilms tumor, SIX1 Q177R hotspot variants were identified at a higher rate in tumor specimens at relapse (11 cases; 13.4%) than in those at diagnosis (4%). For 45 cases that had both diagnostic and relapse specimens, there were 6 cases with SIX1 Q177R at relapse, 3 of which did not have SIX1 Q177R at diagnosis. This finding suggests that this variant is not required for tumor development in some individuals with Wilms tumor.
- TP53 (tumor suppressor gene). Most anaplastic Wilms tumor cases show variants in the TP53 tumor suppressor gene. TP53 may be useful as an unfavorable prognostic marker.
In a study of 118 prospectively identified patients with diffuse anaplastic Wilms tumor registered on the NWTS-5 trial, 57 patients (48%) demonstrated TP53 variants, 13 patients (11%) demonstrated TP53 segmental copy number loss without variants, and 48 patients (41%) lacked both (wild-type TP53 [wtTP53]). All TP53 variants were detected by sequencing alone. Patients with stage III or stage IV disease with wtTP53 had a significantly lower relapse rate and mortality rate than did patients with TP53 abnormalities (P = .00006 and P = .00007, respectively). The TP53 status had no effect on patients with stage I or stage II tumors.
- In-depth analysis of a subset of 39 patients with diffuse anaplastic Wilms tumor showed that 7 patients (18%) were wtTP53. These wtTP53 tumors demonstrated gene expression evidence of p53 pathway activation. Retrospective pathology review of wtTP53 tumors revealed no or very low volume of anaplasia in six of seven tumors. These data support the key role of TP53 loss in the development of anaplasia in Wilms tumor and support its significant clinical influence in patients who have residual anaplastic disease after surgery.
- FBXW7.FBXW7, a ubiquitin ligase component, is an established tumor suppressor gene that has been identified as recurrently altered at low rates in Wilms tumor and other malignancies. Variants of this gene have been associated with epithelial-type tumor histology.; [Level of evidence C1]
- TRIM28.TRIM28 encodes a multidomain protein involved in the regulation of many cellular processes and is an autosomal dominant Wilms tumor predisposition gene. TRIM28 accounts for about 8% of familial Wilms tumor and 2% of unselected Wilms tumor.; [Level of evidence C1]
- A strong association between TRIM28 variants and epithelial Wilms tumor has been observed, and most individuals with a TRIM28 variant have a Wilms tumor of predominantly epithelial histology.; [Level of evidence C1]
- In a cohort of 91 affected individuals from 49 families with Wilms tumor pedigrees, 33 individuals were identified as having constitutional cancer-predisposing variants, 21 of whom had a variant in TRIM28. There was a strong parent-of-origin effect, with all ten evaluable cases having inherited variants that were maternally transmitted.[Level of evidence C1]
- Most TRIM28-altered cases have either frameshift, nonsense, or splice-site variants in one allele combined with LOH in the second allele, leading to loss of TRIM28 protein expression in the tumor. Immunohistochemistry staining for loss of TRIM28 protein expression can be used to identify most patients whose tumors have TRIM28 variants.
- 9q22.3 microdeletion syndrome. Patients with 9q22.3 microdeletion syndrome have an increased risk of Wilms tumor. The chromosomal region with germline deletion includes PTCH1, the gene that is altered in Gorlin syndrome (nevoid basal cell carcinoma syndrome associated with osteosarcoma). 9q22.3 microdeletion syndrome is characterized by the clinical findings of Gorlin syndrome, as well as developmental delay and/or intellectual disability, metopic craniosynostosis, obstructive hydrocephalus, prenatal and postnatal macrosomia, and seizures. Five patients who presented with Wilms tumor in the context of a constitutional 9q22.3 microdeletion have been reported.
- MYCN. Genomic alterations involving the MYCN network (e.g., MYCN, MAX, MGA, NONO) have been reported to occur in 25% to 30% of Wilms tumor cases. Specific genomic alterations associated with the MYCN network include the following:
- MYCN copy number gain was observed in approximately 13% of Wilms tumor cases. MYCN gain was more common in anaplastic cases (7 of 23 cases, 30%) than in nonanaplastic cases (11.2%), and it was associated with poorer relapse-free survival (RFS) and overall survival, independent of histology. MYCN tandem duplication was reported in 11 of 82 (13%) FH Wilms tumor specimens from relapse.
- Germline copy number gain at MYCN has been reported in a bilateral Wilms tumor case, and germline MYCN duplication was also reported for a child with prenatal bilateral nephroblastomatosis and a family history of nephroblastoma.
- Variants at codon 44 (p.P44L) of MYCN are observed in approximately 3% to 4% of Wilms tumor cases at diagnosis and in 8.5% of cases at relapse. In a study of 810 Wilms tumor cases, 24 (3%) had MYCN P44L hotspot variants. RFS was significantly lower (68.6%) in patients with P44L variants than in patients with wild-type MYCN status (87.1%).
- The MYCN interacting protein MAX was altered at codon 60 (R60Q) in 7 of 782 Wilms tumor cases (0.9%). RFS was significantly lower in patients with the MAX R60Q hotspot variant than in patients with wild-type MAX status.
- CTR9. Inactivating CTR9 germline variants were identified in 4 of 36 familial Wilms tumor pedigrees. CTR9, which is located at chromosome 11p15.3, is a key component of the polymerase-associated factor 1 complex (PAF1c), which has multiple roles in RNA polymerase II regulation and is implicated in embryonic organogenesis and maintenance of embryonic stem cell pluripotency.
- REST. Inactivating germline variants in REST (encoding RE1-silencing transcription factor) were identified in four familial Wilms tumor pedigrees. REST is a transcriptional repressor that functions in cellular differentiation and embryonic development. Most REST variants clustered within the portion of REST encoding the DNA-binding domain, and functional analyses showed that these variants compromise REST transcriptional repression. When screened for REST variants, 9 of 519 individuals with Wilms tumor who had no history of relatives with the disease tested positive for the variant; some had parents who also tested positive. These observations indicate that REST is a Wilms tumor predisposition gene associated with approximately 2% of Wilms tumor.
Figure 11 summarizes the genomic landscape of a selected cohort of Wilms tumor patients selected because they experienced relapse despite showing FH. The 75 FH Wilms tumor cases were clustered by unsupervised analysis of gene expression data, resulting in six clusters. Five of six MLLT1-altered tumors with available gene expression data were in cluster 3, and two were accompanied by CTNNB1 variants. This cluster also contained four tumors with a variant or small segment deletion of WT1, all of which also had either a variant of CTNNB1 or small segment deletion or variant of AMER1. It also contained a substantial number of tumors with retention of imprinting of 11p15 (including all MLLT1-altered tumors). The miRNAPG-altered cases clustered together and were mutually exclusive with both MLLT1 and with WT1-, AMER1-, or CTNNB1-altered cases.
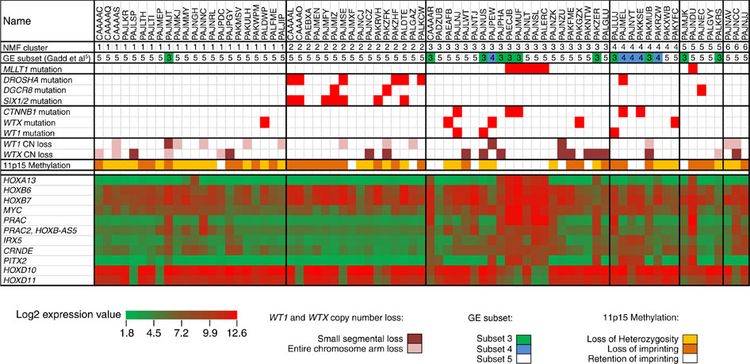 Figure 11. Unsupervised analysis of gene expression data. Non-negative Matrix Factorization (NMF) analysis of 75 FH Wilms tumor resulted in six clusters. Five of six MLLT1 mutant tumors with available gene expression data occurred in NMF cluster 3, and two were accompanied by CTNNB1 mutations. This cluster also contained a substantial number of tumors with retention of imprinting of 11p15 (including all MLLT1-mutant tumors), in contrast to other clusters, where most cases showed 11p15 loss of heterozygosity or retention of imprinting. Almost all miRNAPG-mutated cases were in NMF cluster 2, and most WT1, WTX, and CTNNB1 mutations were in NMF clusters 3 and 4. Copyright © 2015 Perlman, E. J. et al. MLLT1 YEATS domain mutations in clinically distinctive Favourable Histology wilms tumours. Nat. Commun. 6:10013 doi: 10.1038/ncomms10013 (2015). This article is distributed by Nature Publishing Group, a division of Macmillan Publishers Limited under a Creative Commons Attribution 4.0 International License, as described at http://creativecommons.org/licenses/by/4.0/.
Figure 11. Unsupervised analysis of gene expression data. Non-negative Matrix Factorization (NMF) analysis of 75 FH Wilms tumor resulted in six clusters. Five of six MLLT1 mutant tumors with available gene expression data occurred in NMF cluster 3, and two were accompanied by CTNNB1 mutations. This cluster also contained a substantial number of tumors with retention of imprinting of 11p15 (including all MLLT1-mutant tumors), in contrast to other clusters, where most cases showed 11p15 loss of heterozygosity or retention of imprinting. Almost all miRNAPG-mutated cases were in NMF cluster 2, and most WT1, WTX, and CTNNB1 mutations were in NMF clusters 3 and 4. Copyright © 2015 Perlman, E. J. et al. MLLT1 YEATS domain mutations in clinically distinctive Favourable Histology wilms tumours. Nat. Commun. 6:10013 doi: 10.1038/ncomms10013 (2015). This article is distributed by Nature Publishing Group, a division of Macmillan Publishers Limited under a Creative Commons Attribution 4.0 International License, as described at http://creativecommons.org/licenses/by/4.0/.
Genomic alterations in Wilms tumor at relapse
Wilms tumor at relapse appears to maintain most of the genomic alterations present at diagnosis, although there may be changes in the prevalence of alterations in specific genes between diagnosis and relapse. A study from the Children’s Oncology Group presented whole-genome sequencing (WGS) data on relapse tumor specimens from 51 patients and corresponding diagnostic specimens from 45 of these patients. For an additional 31 patients who had relapse specimens available, a targeted sequencing panel was applied. Key findings included the following:
- The prevalence of 1q gain in relapsed Wilms tumor specimens (75%) was higher than that observed for tumors at diagnosis (47%). The increased prevalence of 1q gain at relapse is consistent with its association with poor prognosis and disease progression.
- SIX1 Q177R hotspot variants were identified at a higher rate in tumor specimens at relapse (11 of 82 cases; 13.4%) than in those at diagnosis (4%). For 45 cases with both diagnostic and relapse specimens, there were 6 cases with SIX1 Q177R at relapse, 3 of which did not have SIX1 Q177R at diagnosis. This is consistent with SIX1 Q177R not being an early tumorigenesis event in some cases.
- Genomic alterations in genes associated with the MYCN network were present in approximately 30% of Wilms tumor cases at relapse. The most common MYCN network alterations were MYCN tandem duplication (13%) and MYCN P44L hotspot variants (11%).
Recurrent and refractory Wilms tumors from 56 pediatric patients underwent tumor sequencing in the National Cancer Institute–Children's Oncology Group (NCI-COG) Pediatric MATCH trial. This process revealed genomic alterations that were considered actionable for treatment in MATCH study arms in 6 of 56 tumors (10.7%). BRCA2 variants were found in 2 of 56 tumors (3.6%).
Genomic alterations in adults with Wilms tumor
Wilms tumor in patients older than 16 years is rare, with an incidence rate of less than 0.2 cases per 1 million people per year. As a result, there are limited data available describing the genomic alterations that are observed in adults with Wilms tumor.
A study of 14 patients with a Wilms tumor diagnosis who were older than 16 years (range, 17–46 years; median age, 31 years) evaluated exonic variants for 1,425 cancer-related genes.
- Five patients (36%) harbored BRAF V600E variants. While BRAF V600E variants are extremely uncommon in pediatric Wilms tumor, they are present in 90% of metanephric adenomas of the kidney, a typically benign condition arising almost exclusively in adults.
- All five adult cases of Wilms tumor with BRAF V600E had better-differentiated areas identical to metanephric adenoma adjacent to areas consistent in appearance with epithelial Wilms tumor.
- Two of the five cases with BRAF V600E variants had TERT promoter variants in addition to BRAF variants.
- ASXL1 variants were observed in 4 of 14 cases, including 1 of 5 cases with BRAF V600E variants and 3 of 9 cases without BRAF V600E variants. ASXL1 variants are not common in pediatric Wilms tumor (approximately 2% of cases).
- For the nine tumors that did not have BRAF variants, some had genomic alterations associated with Wilms tumor in children (e.g., 1q gain and variants in WT1 [n = 2]).
Another report described renal tumors that had histological overlap between metanephric adenoma and epithelial Wilms tumor. While most epithelial Wilms tumors (five of nine) with areas resembling metanephric adenoma were negative for BRAF V600E variants, four cases were positive for the BRAF V600E variant. Two of the cases with BRAF V600E variants occurred in children (aged 3 years and 6 years), and the other two cases occurred in adults.
For information about the treatment of Wilms tumor, see Wilms Tumor and Other Childhood Kidney Tumors Treatment.
Renal Cell Carcinoma
Molecular features of renal cell carcinoma
Translocation-positive carcinomas of the kidney are recognized as a distinct form of renal cell carcinoma (RCC) and may be the most common form of RCC in children, accounting for 40% to 50% of pediatric RCC.; [Level of evidence C1] In a Children's Oncology Group (COG) prospective clinical trial of 120 childhood and adolescent patients with RCC, nearly one-half of patients had translocation-positive RCC. These carcinomas are characterized by translocations involving the TFE3 gene located on Xp11.2. The TFE3 gene may partner with one of the following genes:
- ASPSCR in t(X;17)(p11.2;q25).
- PRCC in t(X;1)(p11.2;q21).
- SFPQ in t(X;1)(p11.2;p34).
- NONO in inv(X;p11.2;q12).
- CLTC in t(X;17)(p11;q23).
- VCP in t(x;9)(p11.23;p13.3).
In a single-institution investigation, molecular data from 22 patients with translocation-positive RCCs were pooled with previously published data. Investigators found that certain copy-number variations were associated with disease aggressiveness in patients with translocation-positive RCCs. Tumors bearing 9p loss, 17q gain, or a genetically high burden of copy-number variations were associated with poor survival in these patients. Three pediatric patients who had an indolent disease course were included in the study and were found to have lower copy-number burdens, which supports the less-aggressive disease course in these patients, as compared with adult patients.[Level of evidence C1]
Another less-common translocation subtype, t(6;11)(p21;q12), involving a TFEB gene fusion, induces overexpression of TFEB. The translocations involving TFE3 and TFEB induce overexpression of these proteins, which can be identified by immunohistochemistry.
Previous exposure to chemotherapy is the only known risk factor for the development of Xp11 translocation RCCs. In one study, the postchemotherapy interval ranged from 4 to 13 years. All reported patients received either a DNA topoisomerase II inhibitor or an alkylating agent.
Controversy exists as to the biological behavior of translocation RCC in children and young adults. Whereas some series have suggested a good prognosis when translocation-positive RCC is treated with surgery alone despite presenting at a more advanced stage (III/IV), a meta-analysis reported that these patients have poorer outcomes. The outcomes for these patients are being studied in the ongoing COG AREN03B2 (NCT00898365) biology and classification study. Vascular endothelial growth factor receptor–targeted therapies and mammalian target of rapamycin (mTOR) inhibitors seem to be active in Xp11 translocation metastatic RCC. Recurrences have been reported 20 to 30 years after initial resection of the translocation-associated RCC.
Diagnosis of Xp11 translocation RCC needs to be confirmed by a molecular genetic approach, rather than using TFE3 immunohistochemistry alone, because reported cases have lacked the translocation.
There is a rare subset of RCC cases that is positive for TFE3 and lack a TFE3 translocation, showing an ALK translocation instead. This subset of cases represents a newly recognized subgroup within RCC that is estimated to involve 15% to 20% of unclassified pediatric RCC. In the eight reported cases in children aged 6 to 16 years, the following was observed:
- ALK was fused to VCL in a t(2;10)(p23;q22) translocation (n = 3). The VCL translocation cases all occurred in children with the sickle cell trait, whereas none of the TPM3 translocation cases did.
- ALK was fused to TPM3 (n = 3).
- ALK was fused to HOOK1 on 1p32 (n = 1).
- t(1;2) translocation fusing ALK and TPM3 (n = 1).
For information about the treatment of renal cell carcinoma, see Wilms Tumor and Other Childhood Kidney Tumors Treatment.
Rhabdoid Tumors of the Kidney
Molecular features of rhabdoid tumors of the kidney
Independent of their anatomical locations, rhabdoid tumors have a common genetic abnormality—loss of function of the SMARCB1 gene located at chromosome 22q11.2 (>95% of tumors). The following text refers to rhabdoid tumors without regard to their primary site. SMARCB1 encodes a component of the SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complex that has an important role in controlling gene transcription. Loss of function occurs by deletions that lead to loss of part or all of the SMARCB1 gene and by variants that are commonly frameshift or nonsense variants that lead to premature truncation of the SMARCB1 protein. A common pathway for achieving complete loss of SMARCB1 function is the combination of a SMARCB1 variant or partial/complete gene deletion for one SMARCB1 allele in conjunction with uniparental disomy for the chromosomal region containing SMARCB1 with loss of part or all of the parental chromosome that has a wild-type SMARCB1 allele. A small percentage of rhabdoid tumors are caused by alterations in SMARCA4, which is the primary ATPase in the SWI/SNF complex. Exome sequencing of 35 cases of rhabdoid tumor identified a very low variant rate, with no genes having recurring variants other than SMARCB1, which appeared to contribute to tumorigenesis.
Germline variants of SMARCB1 have been documented in patients with one or more primary tumors of the brain and/or kidney, consistent with a genetic predisposition to the development of rhabdoid tumors. Approximately one-third of patients with rhabdoid tumors have germline SMARCB1 alterations. In most cases, the variants are de novo and not inherited. The median age at diagnosis of children with rhabdoid tumors and a germline variant or deletion is younger (6 months) than that of children with apparently sporadic disease (18 months). Early-onset, multifocal disease and familial cases with the presence of SMARCB1 strongly support the possibility of rhabdoid tumor predisposition syndrome, type 1.
In a study of 100 patients with rhabdoid tumors of the brain, kidney, or soft tissues, 35 were found to have a germline SMARCB1 abnormality. These abnormalities included point and frameshift variants, intragenic deletions and duplications, and larger deletions. Nine cases demonstrated parent-to-child transmission of an altered copy of SMARCB1. In eight of the nine cases, one or more family members were also diagnosed with rhabdoid tumor or schwannoma. Two of the eight families presented with multiple affected children, consistent with gonadal mosaicism. It appears that patients with germline variants may have the worst prognosis.
Rarely, extracranial rhabdoid tumors can harbor the alternative inactivation of SMARCA4 instead of SMARCB1. In a series of 12 patients diagnosed with extracranial rhabdoid tumors with SMARCA4 inactivation, 4 cases occurred in the kidney. All four cases had germline alteration of SMARCA4. The cases of SMARCA4 inactivation were comparable to the extracranial rhabdoid tumors with SMARCB1 inactivation on a clinical, pathological, and genomic level. Using DNA methylation and transcriptomics-based tumor classification, the extracranial rhabdoid tumors with SMARCA4 inactivation display molecular features intermediate between small cell carcinoma of the ovary, hypercalcemic type (driven by SMARCA4 alterations), and extracranial rhabdoid tumors with SMARCB1 inactivations. Extracranial rhabdoid tumors with SMARCA4 inactivation display concomitant lack of SMARCA4 (BRG1) and SMARCA2 (BRM) expression at the protein level, similar to what is seen in small cell carcinoma of the ovary, hypercalcemic type. These results help to expand the similarities and differences between these three tumor types within the rhabdoid tumor spectrum.
For information about the treatment of rhabdoid tumor of the kidney, see Wilms Tumor and Other Childhood Kidney Tumors Treatment.
Clear Cell Sarcoma of the Kidney
Molecular features of clear cell sarcoma of the kidney
The molecular background of clear cell sarcoma of the kidney is poorly understood because of its rarity and lack of experimental models. However, several molecular features of clear cell sarcoma of the kidney have been described, including the following:
- Internal tandem duplications in exon 15 of the BCOR gene (BCL6 corepressor) have been reported in 90% of cases of clear cell sarcoma of the kidney, with a smaller subset harboring YWHAE::NUTM2B, YWHAE::NUTM2E, or BCOR::CCNB3 gene fusions. All of these genetic abnormalities result in a transcriptional signature characterized by high BCOR mRNA expression.
- Diffuse strong immunoreactivity for BCOR is highly sensitive and specific for the diagnosis of clear cell sarcoma of the kidney. One series evaluated 79 neoplasms, including Wilms tumors, congenital mesoblastic nephromas, clear cell sarcoma of the kidney, metanephric stromal tumors, rhabdoid tumors of the kidney, renal primitive neuroectodermal tumor (PNET), and sclerosing epithelioid fibrosarcomas. All of the clear cell sarcoma of the kidney samples that were tested demonstrated diffuse, strong nuclear labeling for BCOR. Most of the other pediatric renal neoplasms were completely negative for BCOR.
For information about the treatment of clear cell tumor of the kidney, see Wilms Tumor and Other Childhood Kidney Tumors Treatment.
References
- Coorens THH, Treger TD, Al-Saadi R, et al.: Embryonal precursors of Wilms tumor. Science 366 (6470): 1247-1251, 2019.
- Gadd S, Huff V, Walz AL, et al.: A Children's Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet 49 (10): 1487-1494, 2017.
- Wegert J, Wittmann S, Leuschner I, et al.: WTX inactivation is a frequent, but late event in Wilms tumors without apparent clinical impact. Genes Chromosomes Cancer 48 (12): 1102-11, 2009.
- Ruteshouser EC, Robinson SM, Huff V: Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer 47 (6): 461-70, 2008.
- Walz AL, Ooms A, Gadd S, et al.: Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 27 (2): 286-97, 2015.
- Wegert J, Ishaque N, Vardapour R, et al.: Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell 27 (2): 298-311, 2015.
- Rakheja D, Chen KS, Liu Y, et al.: Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat Commun 2: 4802, 2014.
- Torrezan GT, Ferreira EN, Nakahata AM, et al.: Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour. Nat Commun 5: 4039, 2014.
- Dome JS, Huff V: Wilms Tumor Predisposition. In: Adam MP, Feldman J, Mirzaa GM, et al., eds.: GeneReviews. University of Washington, Seattle, 1993-2024, pp. Available online. Last accessed August 16, 2022.
- Mahamdallie SS, Hanks S, Karlin KL, et al.: Mutations in the transcriptional repressor REST predispose to Wilms tumor. Nat Genet 47 (12): 1471-4, 2015.
- Hanks S, Perdeaux ER, Seal S, et al.: Germline mutations in the PAF1 complex gene CTR9 predispose to Wilms tumour. Nat Commun 5: 4398, 2014.
- Huff V: Wilms tumor genetics. Am J Med Genet 79 (4): 260-7, 1998.
- Scott RH, Murray A, Baskcomb L, et al.: Stratification of Wilms tumor by genetic and epigenetic analysis. Oncotarget 3 (3): 327-35, 2012.
- Corbin M, de Reyniès A, Rickman DS, et al.: WNT/beta-catenin pathway activation in Wilms tumors: a unifying mechanism with multiple entries? Genes Chromosomes Cancer 48 (9): 816-27, 2009.
- Maiti S, Alam R, Amos CI, et al.: Frequent association of beta-catenin and WT1 mutations in Wilms tumors. Cancer Res 60 (22): 6288-92, 2000.
- Gadd S, Huff V, Huang CC, et al.: Clinically relevant subsets identified by gene expression patterns support a revised ontogenic model of Wilms tumor: a Children's Oncology Group Study. Neoplasia 14 (8): 742-56, 2012.
- Breslow NE, Beckwith JB, Perlman EJ, et al.: Age distributions, birth weights, nephrogenic rests, and heterogeneity in the pathogenesis of Wilms tumor. Pediatr Blood Cancer 47 (3): 260-7, 2006.
- Fukuzawa R, Heathcott RW, More HE, et al.: Sequential WT1 and CTNNB1 mutations and alterations of beta-catenin localisation in intralobar nephrogenic rests and associated Wilms tumours: two case studies. J Clin Pathol 60 (9): 1013-6, 2007.
- Perlman EJ, Gadd S, Arold ST, et al.: MLLT1 YEATS domain mutations in clinically distinctive Favourable Histology Wilms tumours. Nat Commun 6: 10013, 2015.
- Diller L, Ghahremani M, Morgan J, et al.: Constitutional WT1 mutations in Wilms' tumor patients. J Clin Oncol 16 (11): 3634-40, 1998.
- Little SE, Hanks SP, King-Underwood L, et al.: Frequency and heritability of WT1 mutations in nonsyndromic Wilms' tumor patients: a UK Children's Cancer Study Group Study. J Clin Oncol 22 (20): 4140-6, 2004.
- Perlman EJ, Grundy PE, Anderson JR, et al.: WT1 mutation and 11P15 loss of heterozygosity predict relapse in very low-risk wilms tumors treated with surgery alone: a children's oncology group study. J Clin Oncol 29 (6): 698-703, 2011.
- Pelletier J, Bruening W, Kashtan CE, et al.: Germline mutations in the Wilms' tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 67 (2): 437-47, 1991.
- Barbosa AS, Hadjiathanasiou CG, Theodoridis C, et al.: The same mutation affecting the splicing of WT1 gene is present on Frasier syndrome patients with or without Wilms' tumor. Hum Mutat 13 (2): 146-53, 1999.
- Lipska BS, Ranchin B, Iatropoulos P, et al.: Genotype-phenotype associations in WT1 glomerulopathy. Kidney Int 85 (5): 1169-78, 2014.
- Lehnhardt A, Karnatz C, Ahlenstiel-Grunow T, et al.: Clinical and molecular characterization of patients with heterozygous mutations in wilms tumor suppressor gene 1. Clin J Am Soc Nephrol 10 (5): 825-31, 2015.
- Scott RH, Stiller CA, Walker L, et al.: Syndromes and constitutional chromosomal abnormalities associated with Wilms tumour. J Med Genet 43 (9): 705-15, 2006.
- Marakhonov AV, Vasilyeva TA, Voskresenskaya AA, et al.: LMO2 gene deletions significantly worsen the prognosis of Wilms' tumor development in patients with WAGR syndrome. Hum Mol Genet 28 (19): 3323-3326, 2019.
- Green DM, Breslow NE, Beckwith JB, et al.: Screening of children with hemihypertrophy, aniridia, and Beckwith-Wiedemann syndrome in patients with Wilms tumor: a report from the National Wilms Tumor Study. Med Pediatr Oncol 21 (3): 188-92, 1993.
- Scott RH, Walker L, Olsen ØE, et al.: Surveillance for Wilms tumour in at-risk children: pragmatic recommendations for best practice. Arch Dis Child 91 (12): 995-9, 2006.
- Breslow NE, Norris R, Norkool PA, et al.: Characteristics and outcomes of children with the Wilms tumor-Aniridia syndrome: a report from the National Wilms Tumor Study Group. J Clin Oncol 21 (24): 4579-85, 2003.
- Scott RH, Douglas J, Baskcomb L, et al.: Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nat Genet 40 (11): 1329-34, 2008.
- Barbaux S, Niaudet P, Gubler MC, et al.: Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet 17 (4): 467-70, 1997.
- Koesters R, Ridder R, Kopp-Schneider A, et al.: Mutational activation of the beta-catenin proto-oncogene is a common event in the development of Wilms' tumors. Cancer Res 59 (16): 3880-2, 1999.
- Koesters R, Niggli F, von Knebel Doeberitz M, et al.: Nuclear accumulation of beta-catenin protein in Wilms' tumours. J Pathol 199 (1): 68-76, 2003.
- Major MB, Camp ND, Berndt JD, et al.: Wilms tumor suppressor WTX negatively regulates WNT/beta-catenin signaling. Science 316 (5827): 1043-6, 2007.
- Rivera MN, Kim WJ, Wells J, et al.: An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science 315 (5812): 642-5, 2007.
- Fukuzawa R, Anaka MR, Weeks RJ, et al.: Canonical WNT signalling determines lineage specificity in Wilms tumour. Oncogene 28 (8): 1063-75, 2009.
- Jenkins ZA, van Kogelenberg M, Morgan T, et al.: Germline mutations in WTX cause a sclerosing skeletal dysplasia but do not predispose to tumorigenesis. Nat Genet 41 (1): 95-100, 2009.
- Grohmann A, Tanneberger K, Alzner A, et al.: AMER1 regulates the distribution of the tumor suppressor APC between microtubules and the plasma membrane. J Cell Sci 120 (Pt 21): 3738-47, 2007.
- DeBaun MR, Siegel MJ, Choyke PL: Nephromegaly in infancy and early childhood: a risk factor for Wilms tumor in Beckwith-Wiedemann syndrome. J Pediatr 132 (3 Pt 1): 401-4, 1998.
- DeBaun MR, Tucker MA: Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J Pediatr 132 (3 Pt 1): 398-400, 1998.
- Breslow N, Olshan A, Beckwith JB, et al.: Epidemiology of Wilms tumor. Med Pediatr Oncol 21 (3): 172-81, 1993.
- Satoh Y, Nakadate H, Nakagawachi T, et al.: Genetic and epigenetic alterations on the short arm of chromosome 11 are involved in a majority of sporadic Wilms' tumours. Br J Cancer 95 (4): 541-7, 2006.
- Algar EM, St Heaps L, Darmanian A, et al.: Paternally inherited submicroscopic duplication at 11p15.5 implicates insulin-like growth factor II in overgrowth and Wilms' tumorigenesis. Cancer Res 67 (5): 2360-5, 2007.
- Lennerz JK, Timmerman RJ, Grange DK, et al.: Addition of H19 'loss of methylation testing' for Beckwith-Wiedemann syndrome (BWS) increases the diagnostic yield. J Mol Diagn 12 (5): 576-88, 2010.
- Mussa A, Molinatto C, Baldassarre G, et al.: Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J Pediatr 176: 142-149.e1, 2016.
- Bliek J, Gicquel C, Maas S, et al.: Epigenotyping as a tool for the prediction of tumor risk and tumor type in patients with Beckwith-Wiedemann syndrome (BWS). J Pediatr 145 (6): 796-9, 2004.
- Rump P, Zeegers MP, van Essen AJ: Tumor risk in Beckwith-Wiedemann syndrome: A review and meta-analysis. Am J Med Genet A 136 (1): 95-104, 2005.
- Brioude F, Lacoste A, Netchine I, et al.: Beckwith-Wiedemann syndrome: growth pattern and tumor risk according to molecular mechanism, and guidelines for tumor surveillance. Horm Res Paediatr 80 (6): 457-65, 2013.
- Bjornsson HT, Brown LJ, Fallin MD, et al.: Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J Natl Cancer Inst 99 (16): 1270-3, 2007.
- Fukuzawa R, Breslow NE, Morison IM, et al.: Epigenetic differences between Wilms' tumours in white and east-Asian children. Lancet 363 (9407): 446-51, 2004.
- Gratias EJ, Dome JS, Jennings LJ, et al.: Association of Chromosome 1q Gain With Inferior Survival in Favorable-Histology Wilms Tumor: A Report From the Children's Oncology Group. J Clin Oncol 34 (26): 3189-94, 2016.
- Chagtai T, Zill C, Dainese L, et al.: Gain of 1q As a Prognostic Biomarker in Wilms Tumors (WTs) Treated With Preoperative Chemotherapy in the International Society of Paediatric Oncology (SIOP) WT 2001 Trial: A SIOP Renal Tumours Biology Consortium Study. J Clin Oncol 34 (26): 3195-203, 2016.
- Gadd S, Huff V, Skol AD, et al.: Genetic changes associated with relapse in favorable histology Wilms tumor: A Children's Oncology Group AREN03B2 study. Cell Rep Med 3 (6): 100644, 2022.
- Grundy PE, Breslow NE, Li S, et al.: Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol 23 (29): 7312-21, 2005.
- Messahel B, Williams R, Ridolfi A, et al.: Allele loss at 16q defines poorer prognosis Wilms tumour irrespective of treatment approach in the UKW1-3 clinical trials: a Children's Cancer and Leukaemia Group (CCLG) Study. Eur J Cancer 45 (5): 819-26, 2009.
- Spreafico F, Gamba B, Mariani L, et al.: Loss of heterozygosity analysis at different chromosome regions in Wilms tumor confirms 1p allelic loss as a marker of worse prognosis: a study from the Italian Association of Pediatric Hematology and Oncology. J Urol 189 (1): 260-6, 2013.
- Gratias EJ, Jennings LJ, Anderson JR, et al.: Gain of 1q is associated with inferior event-free and overall survival in patients with favorable histology Wilms tumor: a report from the Children's Oncology Group. Cancer 119 (21): 3887-94, 2013.
- Hohenstein P, Pritchard-Jones K, Charlton J: The yin and yang of kidney development and Wilms' tumors. Genes Dev 29 (5): 467-82, 2015.
- Foulkes WD, Priest JR, Duchaine TF: DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer 14 (10): 662-72, 2014.
- Wu MK, Sabbaghian N, Xu B, et al.: Biallelic DICER1 mutations occur in Wilms tumours. J Pathol 230 (2): 154-64, 2013.
- Palculict TB, Ruteshouser EC, Fan Y, et al.: Identification of germline DICER1 mutations and loss of heterozygosity in familial Wilms tumour. J Med Genet 53 (6): 385-8, 2016.
- Astuti D, Morris MR, Cooper WN, et al.: Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat Genet 44 (3): 277-84, 2012.
- Chang HM, Triboulet R, Thornton JE, et al.: A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature 497 (7448): 244-8, 2013.
- Hol JA, Kuiper RP, van Dijk F, et al.: Prevalence of (Epi)genetic Predisposing Factors in a 5-Year Unselected National Wilms Tumor Cohort: A Comprehensive Clinical and Genomic Characterization. J Clin Oncol 40 (17): 1892-1902, 2022.
- Alessandri JL, Cuillier F, Ramful D, et al.: Perlman syndrome: report, prenatal findings and review. Am J Med Genet A 146A (19): 2532-7, 2008.
- Bardeesy N, Falkoff D, Petruzzi MJ, et al.: Anaplastic Wilms' tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nat Genet 7 (1): 91-7, 1994.
- el Bahtimi R, Hazen-Martin DJ, Re GG, et al.: Immunophenotype, mRNA expression, and gene structure of p53 in Wilms' tumors. Mod Pathol 9 (3): 238-44, 1996.
- Wallkamm V, Dörlich R, Rahm K, et al.: Live imaging of Xwnt5A-ROR2 complexes. PLoS One 9 (10): e109428, 2014.
- Ooms AH, Gadd S, Gerhard DS, et al.: Significance of TP53 Mutation in Wilms Tumors with Diffuse Anaplasia: A Report from the Children's Oncology Group. Clin Cancer Res 22 (22): 5582-5591, 2016.
- Williams RD, Al-Saadi R, Chagtai T, et al.: Subtype-specific FBXW7 mutation and MYCN copy number gain in Wilms' tumor. Clin Cancer Res 16 (7): 2036-45, 2010.
- Mahamdallie S, Yost S, Poyastro-Pearson E, et al.: Identification of new Wilms tumour predisposition genes: an exome sequencing study. Lancet Child Adolesc Health 3 (5): 322-331, 2019.
- Armstrong AE, Gadd S, Huff V, et al.: A unique subset of low-risk Wilms tumors is characterized by loss of function of TRIM28 (KAP1), a gene critical in early renal development: A Children's Oncology Group study. PLoS One 13 (12): e0208936, 2018.
- Halliday BJ, Fukuzawa R, Markie DM, et al.: Germline mutations and somatic inactivation of TRIM28 in Wilms tumour. PLoS Genet 14 (6): e1007399, 2018.
- Diets IJ, Hoyer J, Ekici AB, et al.: TRIM28 haploinsufficiency predisposes to Wilms tumor. Int J Cancer 145 (4): 941-951, 2019.
- Hol JA, Diets IJ, de Krijger RR, et al.: TRIM28 variants and Wilms' tumour predisposition. J Pathol 254 (4): 494-504, 2021.
- Isidor B, Bourdeaut F, Lafon D, et al.: Wilms' tumor in patients with 9q22.3 microdeletion syndrome suggests a role for PTCH1 in nephroblastomas. Eur J Hum Genet 21 (7): 784-7, 2013.
- Garavelli L, Piemontese MR, Cavazza A, et al.: Multiple tumor types including leiomyoma and Wilms tumor in a patient with Gorlin syndrome due to 9q22.3 microdeletion encompassing the PTCH1 and FANC-C loci. Am J Med Genet A 161A (11): 2894-901, 2013.
- Cajaiba MM, Bale AE, Alvarez-Franco M, et al.: Rhabdomyosarcoma, Wilms tumor, and deletion of the patched gene in Gorlin syndrome. Nat Clin Pract Oncol 3 (10): 575-80, 2006.
- Williams RD, Chagtai T, Alcaide-German M, et al.: Multiple mechanisms of MYCN dysregulation in Wilms tumour. Oncotarget 6 (9): 7232-43, 2015.
- Fievet A, Belaud-Rotureau MA, Dugay F, et al.: Involvement of germline DDX1-MYCN duplication in inherited nephroblastoma. Eur J Med Genet 56 (12): 643-7, 2013.
- Jiménez Martín O, Schlosser A, Furtwängler R, et al.: MYCN and MAX alterations in Wilms tumor and identification of novel N-MYC interaction partners as biomarker candidates. Cancer Cell Int 21 (1): 555, 2021.
- Martins AG, Pinto AT, Domingues R, et al.: Identification of a novel CTR9 germline mutation in a family with Wilms tumor. Eur J Med Genet 61 (5): 294-299, 2018.
- Parsons DW, Janeway KA, Patton DR, et al.: Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients With Refractory Cancers in the National Cancer Institute-Children's Oncology Group Pediatric MATCH Trial. J Clin Oncol 40 (20): 2224-2234, 2022.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 15, 2023.
- Argani P, Tickoo SK, Matoso A, et al.: Adult Wilms Tumor: Genetic Evidence of Origin of a Subset of Cases From Metanephric Adenoma. Am J Surg Pathol 46 (7): 988-999, 2022.
- Choueiri TK, Cheville J, Palescandolo E, et al.: BRAF mutations in metanephric adenoma of the kidney. Eur Urol 62 (5): 917-22, 2012.
- Wobker SE, Matoso A, Pratilas CA, et al.: Metanephric Adenoma-Epithelial Wilms Tumor Overlap Lesions: An Analysis of BRAF Status. Am J Surg Pathol 43 (9): 1157-1169, 2019.
- Geller JI, Dome JS: Local lymph node involvement does not predict poor outcome in pediatric renal cell carcinoma. Cancer 101 (7): 1575-83, 2004.
- van der Beek JN, Hol JA, Coulomb-l'Hermine A, et al.: Characteristics and outcome of pediatric renal cell carcinoma patients registered in the International Society of Pediatric Oncology (SIOP) 93-01, 2001 and UK-IMPORT database: A report of the SIOP-Renal Tumor Study Group. Int J Cancer 148 (11): 2724-2735, 2021.
- Geller JI, Ehrlich PF, Cost NG, et al.: Characterization of adolescent and pediatric renal cell carcinoma: A report from the Children's Oncology Group study AREN03B2. Cancer 121 (14): 2457-64, 2015.
- Ambalavanan M, Geller JI: Treatment of advanced pediatric renal cell carcinoma. Pediatr Blood Cancer 66 (8): e27766, 2019.
- Ge Y, Lin X, Zhang Q, et al.: Xp11.2 Translocation Renal Cell Carcinoma With TFE3 Rearrangement: Distinct Morphological Features and Prognosis With Different Fusion Partners. Front Oncol 11: 784993, 2021.
- Marcon J, DiNatale RG, Sanchez A, et al.: Comprehensive Genomic Analysis of Translocation Renal Cell Carcinoma Reveals Copy-Number Variations as Drivers of Disease Progression. Clin Cancer Res 26 (14): 3629-3640, 2020.
- Argani P, Hicks J, De Marzo AM, et al.: Xp11 translocation renal cell carcinoma (RCC): extended immunohistochemical profile emphasizing novel RCC markers. Am J Surg Pathol 34 (9): 1295-303, 2010.
- Argani P, Laé M, Ballard ET, et al.: Translocation carcinomas of the kidney after chemotherapy in childhood. J Clin Oncol 24 (10): 1529-34, 2006.
- Ramphal R, Pappo A, Zielenska M, et al.: Pediatric renal cell carcinoma: clinical, pathologic, and molecular abnormalities associated with the members of the mit transcription factor family. Am J Clin Pathol 126 (3): 349-64, 2006.
- Geller JI, Argani P, Adeniran A, et al.: Translocation renal cell carcinoma: lack of negative impact due to lymph node spread. Cancer 112 (7): 1607-16, 2008.
- Camparo P, Vasiliu V, Molinie V, et al.: Renal translocation carcinomas: clinicopathologic, immunohistochemical, and gene expression profiling analysis of 31 cases with a review of the literature. Am J Surg Pathol 32 (5): 656-70, 2008.
- Qiu Rao, Bing Guan, Zhou XJ: Xp11.2 Translocation renal cell carcinomas have a poorer prognosis than non-Xp11.2 translocation carcinomas in children and young adults: a meta-analysis. Int J Surg Pathol 18 (6): 458-64, 2010.
- Malouf GG, Camparo P, Oudard S, et al.: Targeted agents in metastatic Xp11 translocation/TFE3 gene fusion renal cell carcinoma (RCC): a report from the Juvenile RCC Network. Ann Oncol 21 (9): 1834-8, 2010.
- Rais-Bahrami S, Drabick JJ, De Marzo AM, et al.: Xp11 translocation renal cell carcinoma: delayed but massive and lethal metastases of a chemotherapy-associated secondary malignancy. Urology 70 (1): 178.e3-6, 2007.
- Thorner PS, Shago M, Marrano P, et al.: TFE3-positive renal cell carcinomas are not always Xp11 translocation carcinomas: Report of a case with a TPM3-ALK translocation. Pathol Res Pract 212 (10): 937-942, 2016.
- Cajaiba MM, Jennings LJ, Rohan SM, et al.: ALK-rearranged renal cell carcinomas in children. Genes Chromosomes Cancer 55 (5): 442-51, 2016.
- Smith NE, Deyrup AT, Mariño-Enriquez A, et al.: VCL-ALK renal cell carcinoma in children with sickle-cell trait: the eighth sickle-cell nephropathy? Am J Surg Pathol 38 (6): 858-63, 2014.
- Cajaiba MM, Jennings LJ, George D, et al.: Expanding the spectrum of ALK-rearranged renal cell carcinomas in children: Identification of a novel HOOK1-ALK fusion transcript. Genes Chromosomes Cancer 55 (10): 814-7, 2016.
- Versteege I, Sévenet N, Lange J, et al.: Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394 (6689): 203-6, 1998.
- Imbalzano AN, Jones SN: Snf5 tumor suppressor couples chromatin remodeling, checkpoint control, and chromosomal stability. Cancer Cell 7 (4): 294-5, 2005.
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011.
- Haruta M, Arai Y, Okita H, et al.: Frequent breakpoints of focal deletion and uniparental disomy in 22q11.1 or 11.2 segmental duplication region reveal distinct tumorigenesis in rhabdoid tumor of the kidney. Genes Chromosomes Cancer 60 (8): 546-558, 2021.
- Schneppenheim R, Frühwald MC, Gesk S, et al.: Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet 86 (2): 279-84, 2010.
- Hasselblatt M, Gesk S, Oyen F, et al.: Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 35 (6): 933-5, 2011.
- Lee RS, Stewart C, Carter SL, et al.: A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122 (8): 2983-8, 2012.
- Biegel JA, Zhou JY, Rorke LB, et al.: Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59 (1): 74-9, 1999.
- Biegel JA: Molecular genetics of atypical teratoid/rhabdoid tumor. Neurosurg Focus 20 (1): E11, 2006.
- Bourdeaut F, Lequin D, Brugières L, et al.: Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res 17 (1): 31-8, 2011.
- Geller JI, Roth JJ, Biegel JA: Biology and Treatment of Rhabdoid Tumor. Crit Rev Oncog 20 (3-4): 199-216, 2015.
- Janson K, Nedzi LA, David O, et al.: Predisposition to atypical teratoid/rhabdoid tumor due to an inherited INI1 mutation. Pediatr Blood Cancer 47 (3): 279-84, 2006.
- Sévenet N, Sheridan E, Amram D, et al.: Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet 65 (5): 1342-8, 1999.
- Hasselblatt M, Nagel I, Oyen F, et al.: SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol 128 (3): 453-6, 2014.
- Andrianteranagna M, Cyrta J, Masliah-Planchon J, et al.: SMARCA4-deficient rhabdoid tumours show intermediate molecular features between SMARCB1-deficient rhabdoid tumours and small cell carcinomas of the ovary, hypercalcaemic type. J Pathol 255 (1): 1-15, 2021.
- Ueno-Yokohata H, Okita H, Nakasato K, et al.: Consistent in-frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney. Nat Genet 47 (8): 861-3, 2015.
- Argani P, Kao YC, Zhang L, et al.: Primary Renal Sarcomas With BCOR-CCNB3 Gene Fusion: A Report of 2 Cases Showing Histologic Overlap With Clear Cell Sarcoma of Kidney, Suggesting Further Link Between BCOR-related Sarcomas of the Kidney and Soft Tissues. Am J Surg Pathol 41 (12): 1702-1712, 2017.
- Karlsson J, Valind A, Gisselsson D: BCOR internal tandem duplication and YWHAE-NUTM2B/E fusion are mutually exclusive events in clear cell sarcoma of the kidney. Genes Chromosomes Cancer 55 (2): 120-3, 2016.
- Astolfi A, Melchionda F, Perotti D, et al.: Whole transcriptome sequencing identifies BCOR internal tandem duplication as a common feature of clear cell sarcoma of the kidney. Oncotarget 6 (38): 40934-9, 2015.
- Roy A, Kumar V, Zorman B, et al.: Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat Commun 6: 8891, 2015.
- Wong MK, Ng CCY, Kuick CH, et al.: Clear cell sarcomas of the kidney are characterised by BCOR gene abnormalities, including exon 15 internal tandem duplications and BCOR-CCNB3 gene fusion. Histopathology 72 (2): 320-329, 2018.
- Kao YC, Sung YS, Zhang L, et al.: Recurrent BCOR Internal Tandem Duplication and YWHAE-NUTM2B Fusions in Soft Tissue Undifferentiated Round Cell Sarcoma of Infancy: Overlapping Genetic Features With Clear Cell Sarcoma of Kidney. Am J Surg Pathol 40 (8): 1009-20, 2016.
- Argani P, Pawel B, Szabo S, et al.: Diffuse Strong BCOR Immunoreactivity Is a Sensitive and Specific Marker for Clear Cell Sarcoma of the Kidney (CCSK) in Pediatric Renal Neoplasia. Am J Surg Pathol 42 (8): 1128-1131, 2018.
Melanoma
For information about the genomics of childhood melanoma, see the Molecular Features section in Childhood Melanoma Treatment.
For information about the treatment of childhood melanoma, see Childhood Melanoma Treatment.
Thyroid Cancer
For information about the genomics of childhood thyroid cancer, see the Molecular Features section in Childhood Thyroid Cancer Treatment.
For information about the treatment of childhood thyroid cancer, see Childhood Thyroid Cancer Treatment.
Multiple Endocrine Neoplasia Syndromes
For information about the genomics of childhood multiple endocrine neoplasia (MEN) syndromes, see the Clinical Presentation, Diagnostic Evaluation, and Molecular Features section in Childhood Multiple Endocrine Neoplasia (MEN) Syndromes Treatment.
For information about the treatment of childhood MEN syndromes, see Childhood Multiple Endocrine Neoplasia (MEN) Syndromes Treatment.
Latest Updates to This Summary (06/25/2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Leukemias
Added Öfverholm et al. as reference 149 in the Acute Lymphoblastic Leukemia (ALL) section.
Central Nervous System Tumors
Added Das et al. as reference 12 in the Astrocytomas, Other Gliomas, and Glioneuronal/Neuronal Tumors section.
Added text to the Astrocytomas, Other Gliomas, and Glioneuronal/Neuronal Tumors section to state that ROS1 gene fusions have been reported in gliomas occurring in older children and adults. A retrospective meta-analysis that included 40 children older than 1 year revealed that ROS1 gene fusions occurred in diverse glioma histologies, including diffuse high-grade and low-grade gliomas and glioneuronal tumors. Similar to ROS1-altered cases occurring in infants, tumor variants in other known driver genes were rare. However, tumor copy number alterations were more frequent in older children than infants (cited Meredith et al. as reference 21).
Added text to the Astrocytomas, Other Gliomas, and Glioneuronal/Neuronal Tumors section to state that a subset of H3 K27-altered tumors will have a BRAF V600E or FGFR1 co-variant. Added text about the results of a retrospective study that demonstrated a somewhat higher propensity for a thalamic location (cited Auffret et al. as reference 41). Also added text about the results of a separate retrospective study of pediatric and adult patients with H3 K27-altered gliomas that revealed BRAF V600E variants in 5.8% and FGFR1 variants in 10.9% of patients younger than 20 years (cited Williams et al. as reference 42).
Added text to the Astrocytomas, Other Gliomas, and Glioneuronal/Neuronal Tumors section to state that the small minority of patients with diffuse midline gliomas lacking histone H3 variants often show EZHIP overexpression. EZHIP inhibits PRC2 activity, leading to the same loss of H3 K27 trimethylation that is induced by H3 K27M variants (cited Jain et al. as reference 43). Overexpression of EZHIP is likewise observed in posterior fossa type A ependymomas, which also shows loss of H3 K27 methylation (cited Hübner et al. as reference 44).
Added text to the Astrocytomas, Other Gliomas, and Glioneuronal/Neuronal Tumors section about the prevalence, presentation, and genomics of IDH1- and IDH2-altered tumors that occur in the pediatric population.
Langerhans Cell Histiocytosis
Added text to the Langerhans Cell Histiocytosis section about the results of a study that used a mouse model with the BRAF V600E variant under control of Scl or Map17 gene promoters and added additional insights into the biology of neurodegenerative LCH (cited Wilk et al. as reference 16).
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the genomics of childhood cancer. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Childhood Cancer Genomics are:
- William L. Carroll, MD (Laura and Isaac Perlmutter Cancer Center at NYU Langone)
- Michelle Hermiston, MD, PhD (University of California, San Francisco)
- Megan S. Lim, MD, PhD (University of Pennsylvania)
- D. Williams Parsons, MD, PhD (Texas Children's Hospital)
- Jessica Pollard, MD (Dana-Farber/Boston Children's Cancer and Blood Disorders Center)
- Rachel E. Rau, MD (University of Washington School of Medicine, Seatle Children’s)
- Lisa Giulino Roth, MD (Weil Cornell Medical College)
- Malcolm A. Smith, MD, PhD (National Cancer Institute)
- Sarah K. Tasian, MD (Children's Hospital of Philadelphia)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Childhood Cancer Genomics. Bethesda, MD: National Cancer Institute. Updated
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.