Familial Colorectal Cancers - Familial Adenomatous Polyposis (FAP)
What is familial adenomatous polyposis (FAP)?
Each year, about 154,000 people in the United States are diagnosed with colorectal cancer. About 5 out of 100 cases of colorectal cancers may be caused by genetic abnormalities (gene changes or mutations) that run in families (passed down from your parents). One of these genetic abnormalities is familial adenomatous polyposis (FAP). FAP is very rare and causes less than one out of 100 cases of colorectal cancer each year.
Adenomatous polyps are called "pre-cancerous" lesions, and some will likely turn into cancer. People with FAP can have hundreds to thousands of polyps. The more polyps you have, the higher your risk of colorectal cancer. If you have FAP, the average onset age of polyps (when the polyps start) is 16. Having FAP raises your risk of getting colorectal cancer by age 40 unless your colon is removed.
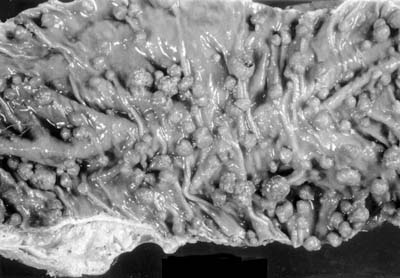
Colon with multiple adenomas, Photo courtesy of Zane Cohen Centre for Digestive Disorders, Mt. Sinai Hospital, Toronto, Canada.
FAP can also raise your risk of other cancers, such as stomach, small intestine, pancreatic, biliary tree, hepatoblastoma, medulloblastoma, and papillary thyroid cancers. FAP can also increase your risk of other non-cancerous issues like:
- Fibromas and cysts on the skin of the face, scalp, arms, and legs.
- Some will have a freckle in the retina of the eye called “congenital hypertrophy of the retinal pigment epithelium” (CHRPE). Someone with FAP will likely have it in both eyes.
- Dental issues, like extra or missing teeth, fused roots, or non-cancerous tumors of the jawbone.
- Osteomas (bony growths) in the jaw.
- Masses in the adrenal glands (small glands on top of your kidneys).
- Desmoid tumors, which are non-cancerous, slow-growing tumors in the abdomen (belly). These can cause health issues when they press against nearby organs and tissues.
If you have any of these health issues, talk to your provider about being tested for FAP.
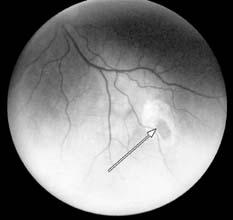
Congenital hypertrophy of the retinal pigment epithelium, Photo courtesy of Zane Cohen Centre for Digestive Disorders, Mt. Sinai Hospital, Toronto, Canada.
Genetic Testing, Screening, and Diagnosing FAP
The gene that causes FAP is called the Adenomatous Polyposis Coli (APC) gene. There are over 300 different mutations (changes) in the APC gene that can cause FAP. Where the mutation is on the gene is often related to the number of polyps, age of onset, and other factors. FAP is an autosomal dominant inherited disorder. This means a child who has a parent with the affected gene has a 50 out of 100 chance of inheriting the mutation. Up to 1 out of 3 people with FAP have "de novo mutations," meaning they have no family history of the mutation.
FAP is diagnosed when you have more than 100 adenomatous polyps found during a colonoscopy or when genetic testing is done in a family with a history of FAP. Once you are diagnosed, other family members should get tested for FAP.
Genetic testing can find abnormalities. There are some things you should think about when it comes to genetic testing:
- Think about how the results will affect you and your family members.
- What will a positive or negative test mean for you or others?
- What are the chances of passing the gene on to your children?
- What challenges you might face with your job and insurance coverage.
You should meet with a genetic counselor if you want testing. They are trained to help you understand the risks and benefits of testing and what to do with the results.
Treatment
Aspirin and some non-steroidal anti-inflammatory drugs (like sulindac and indomethacin) have been shown to shrink or reduce the number of polyps in people with FAP. A clinical trial studied the benefits of a targeted therapy medication called erlotinib with sulindac. Clinical trials are ongoing to find ways to prevent polyp growth.
If you have FAP, you will need your colon removed to prevent getting colon cancer. This surgery is often done when you are in your late teens and don’t have any symptoms of FAP. There are many surgical options. The goal is to remove all colorectal tissue at risk for polyps and to continue being able to pass stool through the anus.
Research about FAP has led to genetic testing for those at-risk, improved screening and surgical options, and clinical trials for the prevention of polyps. Research for treatment of FAP is ongoing. If you have any questions about FAP, you can always ask your providers or genetic specialists.
Resources for More Information
Find a genetic counselor in your area on the National Society of Genetic Counselors website.
Learn more about FAP on the Colorectal Cancer Alliance website.
Learn more about mutations in the APC gene on the FORCE website.